بیماری غشای پایهٔ نازک و سندرم آلپورت

بیماری غشای پایهٔ نازک
thin basement membrane disease
تخمین زده میشود که بیماری غشای پایهٔ نازک یا هماچوری فامیلی خوشخیم 1% از جمعیت عمومی را مبتلا میکند. حدود 50% از مبتلایان به بیماری غشای پایهٔ نازک الگوی انتقال اتوزوم غالب نشان میدهند و 40 تا 50 درصد از بیماران نیز جهشهایی در ژن COL4A3 یا COL4A4 دارند. این جهشها باعث کاهشهای ناچیزی در شبکهٔ α3α4α5 کلاژن نوع IV در غشای پایهٔ گلومرول میشوند. این وضعیت با سندرم آلپورت که در آن شبکههای کلاژن IV از بین رفته یا به شدت تغییر شکل پیدا کردهاند تفاوت دارد اما سبب شده تا بعضی صاحبنظران بیماری غشای پایهٔ نازک را شکل خفیف گلومرولوپاتی ناشی از اختلال کلاژن IV و سندرم آلپورت را شکل شدید آن ببینند. بیماران منحصراً با هماچوری میکروسکوپی تظاهر پیدا میکنند. پروتئینوری وجود ندارد. تشخیص با نمونهبرداری از کلیه مسجل میشود که نازک شدن لایهٔ متراکم (lamina densa) غشای پایهٔ گلومرول، معمولاً به کمتر از 250 نانومتر در بزرگسالان را نشان میدهد. تمایز بیماری غشای پایهٔ نازک کلاسیک از سندرم آلپورت حایز اهمیت است زیرا بیماری غشای پایهٔ نازک برخلاف سندرم آلپورت پیشآگهی کاملاً خوشخیمی دارد.

سندرم آلپورت
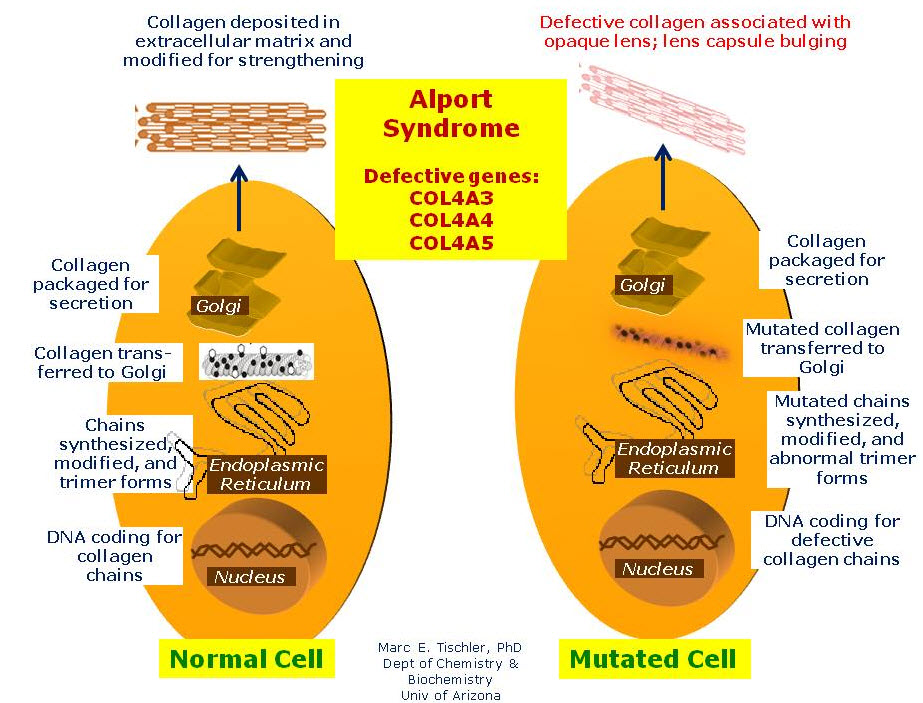
alport syndrome
سندرم آلپورت که با عنوان نفریت ارثی نیز شناخته میشود طیفی از بیماران با جهش در کلاژن IV را در بر میگیرد که با هماچوری تظاهر مییابند. حدود 85% از بیماران یک جهش وابسته به X در COL4A5 دارند که باعث کاهش یا به هم ریختن کلاژن (IV) α3α4α5 و کلاژن (IV) α5α5α6 میشود. ناقلین مؤنث بسته به نوع جهش یا درجهٔ موزائیسیزم از نفوذ (penetrance) متغیر برخوردارند. بیماری اتوزوم مغلوب با جهش در COL4A3 یا COL4A4 که باعث ناهنجاری شبکهٔ (IV) α3α4α5 میشود نیز گزارش شدهاند. بیماران با هماچوری میکروسکوپی، پروتئینوری خفیف، و درجات متغیری از کری حسی ـ عصبی تظاهر مییابند؛ برخی بیماران دچار برجستگی مخروطی شکل عدسی (lenticonus) در کپسول قدامی عدسی میشوند. ندرتاً عقبماندگی ذهنی و لیومیوماتوز با سندرم آلپورت همراهی دارند. بیماران مذکر بیشتر از بیماران مؤنث مبتلا میشوند و به طور شایعتر به سمت گلومرولواسکلروز مزمن و بیماری کلیوی مرحلهٔ نهایی پیشرفت میکنند. در شکل مرتبط با X سندرم آلپورت که در کودکی بروز میکند بیماران مذکر قبل از 30 سالگی به بیماری کلیوی مرحلهٔ نهایی میرسند و در شکل بالغین بیماری کلیوی مرحلهٔ نهایی پس از 30 سالگی رخ میدهد. کری زودرس شدید یا برجستگی مخروطی شکل عدسی حاکی از پیشآگهی وخیمتر میباشند. در نمونهبرداری از پوست 80% بیماران مذکر مبتلا به سندرم آلپورت وابسته به X رنگپذیری منفی برای (IV) α5 در غشای پایهٔ اپیدرم و رنگپذیری ایمنی (immunostainig) موزائیک در 50% بیماران مؤنث مبتلا به سندرم آلپورت وابسته به X دیده میشود. هنگامی که نمونهبرداری از پوست در سندرم آلپورت وابسته به X تشخیصی نباشد (20 تا 50 درصد) و در بیماران مبتلا به سندرم آلپورت اتوزوم مغلوب یا غالب نمونهبرداری از کلیه انجام میشود. در اوایل بیماری، مبتلایان به سندرم آلپورت غشای پایهٔ گلومرولی نازک دارند که در طول زمان به صورت چندین لایهٔ احاطهکنندهٔ فضاهای شفاف ضخیم میگردد؛ این وضعیت را غشای پایهٔ شکافدار (split basement membrane) مینامند. درمان اصلی، کنترل فشارخون سراسری با استفاده از مهارکنندههای سامانهٔ رنین ـ آنیوتانسین میباشد.
گلومرولوپاتی مرتبط با کمخونی سلول داسی، و اسکولیت مرتبط با آنتیبادی سیتوپلاسمی ضدهسته (ANCA)، و نفریت لوپوسی نیز میتوانند با هماچوری تظاهر پیدا کنند.