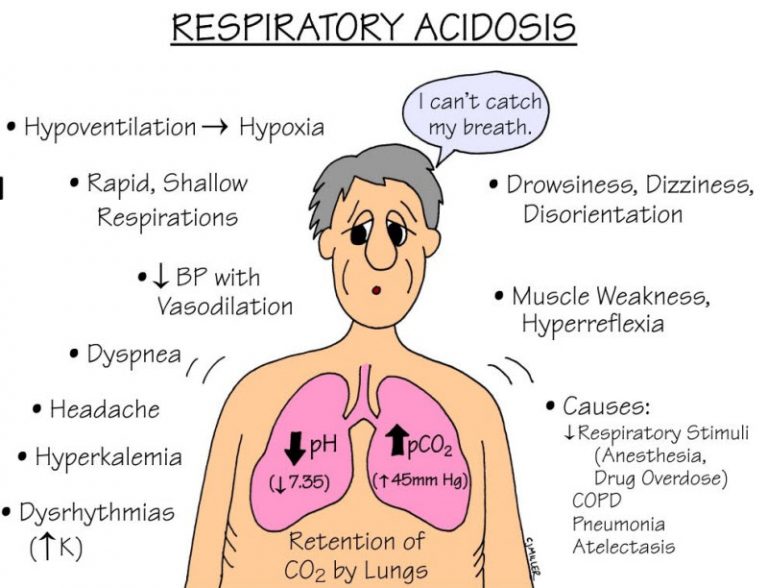
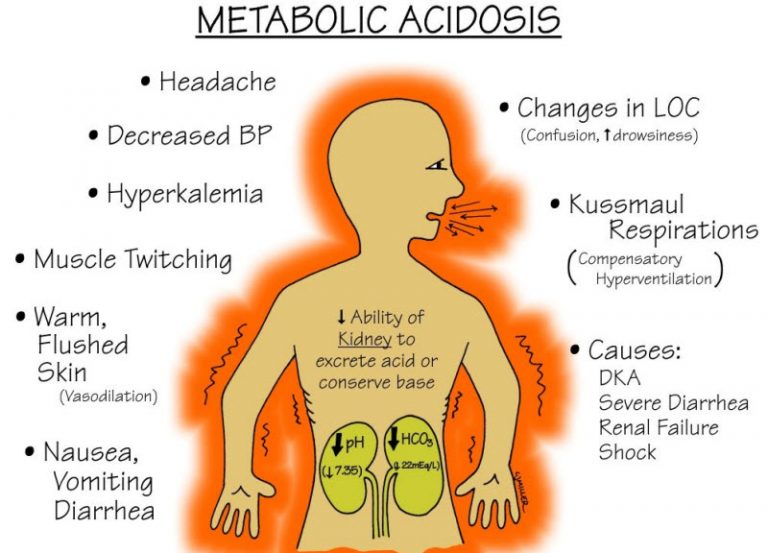
آلکالوز متابولیک چیست؟

دریافت باز (قلیا) یا از دست دادن اسید، غلظت بیکربنات در مایع خارج سلولی را افزایش میدهد. به طور طبیعی افزای غلظت سرمی بیکربنات اضافی اصلاح میشود. بنابراین، تداوم آلکالوز متابولیک حاکی از آن است که مکانیسمهای تنظیمکننده دفع بیکربنات در کلیه، دارای نقصاند. ناتوانی در دفع مقادیر اضافی بیکربنات، هم توسط پاسخهای فیزیولوژیک به کاهش حجم (به خصوص در صورت همراهی هیپرکاپنی و هیپوکالمی با آلکالوز) و هم توسط پاسخهای پاتوفیزیولوژیک (مانند ترشح مقادیر زیاد مینرالوکورتیکوئید به طور خودکار) ایجاد میشود.
شایعترین علت آلکالوز متابولیک، دفع اسید هیدروکلریک معده از طریق استفراغ یا تخلیه مکانیکی است. مصرف داروی مدر (تیازید و داروهای مؤثر بر قوس هنله) معمولاً با آلکالوز متابولیک همراه است. کاهش حجم ناشی از استفراغ و مصرف داروهای مدر بازجذب بیکربنات را در لوله نزدیک تقویت میکند. افزایش معاوضهٔ سدیم ـ هیدروژن در این مکان و درنتیجه افزایش بازجذب آب، منجر به افزایش بازجذب بیکربنات میشود (به فصل 26 مراجعه کنید). کاهش حجم، همچنین منجر به ترشح آلدوسترون میشود. آلدوسترون، ترشح هیدروژن را از نفرون دور و نیز، ترشح پتاسیم را افزایش میدهد. تصحیح آلکالوز تحت این شرایط، نیازمند تجویز کلرید سدیم و پتاسیم است. افزایش مینرالوکورتیکوئیدهای درونزاد یا برونزاد (شکل 7 ـ 28)، با تجویز مایعات تصحیح نمیشود زیرا حجم مایع خارج سلولی افزایش مییابد. تحریک ترشح هیدروژن دیستال، برای محدود کردن دفع بیکربنات و تحریک ترشح پتاسیم کفایت میکند. تصحیح این اختلال، نیازمند حذف مینرالوکورتیکوئید اضافی است. در تمام این اختلالات، ایجاد همزمان هیپوکالمی منجر به باقی ماندن آلکالوز متابولیک میشود.
مصرف بیش از حد قلیا (مانند سندرم شیر قلیا)، از علل نادر الکالوز متابولیک است. این حالت ناشی از اختلال در دفع کلیوی بیکربنات بر اثر نارسایی کلیه در زمینهٔ مصرف زیاد قلیا میباشد. در این مورد، به نظر میرسد هم هیپرکلسمی و هم اضافه بودن ویتامین D در آسیب زدن به کلیه دخیلند. حذف قلیاها، غالباً منجر به برطرف شدن آلکالوز میشود ولی در صورت قابل توجه بودن نفروکلسینوز، عملکرد کلیه در حد پایین باقی میماند.
اندازهگیری غلظت کلر ادرار به برنامهریزی منطقی برای تشخیص و درمان آلکالوز متابولیک کمک میکند. در بیمارانی که از زمان کودکی مبتلا به فشارخون بالا بودهاند و همچنین در آنها آلکالوز وهیپوکالمی دیده میشود و غلظت کلر ادرار پایین است، باید به فکر سندرم لیدل (Liddle ‘ssyndrome) بود. این خصوصیات، مشابه خصوصیات ناشی از اضافی بودن مینرالوکورتیکوئیدها هستند ولی مقادیر رنین و آلدوسترون پایین است (هیپوآلدسترونیسم کاذب). این اختلال به صورت یک اختلال اتوزومی مغلوب به ارث میرسد و جهش در ژن ENaC، منجر به حذف ناحیهٔ پایانهٔ C (C-terminal) پروتئین میگردد. این وضعیت، منجربه کاهش تخریب و افزایش تعداد کانالهای سدیم در غشای مجرایی سلولهای اصلی مجرای جمعکننده میگردد (فصل 26). خصوصیات بالینی، نتیجهٔ افزایش بازجذب نمک و درنتیجه، افزایش حجم و افزایش ترشح پتاسیم و پروتون از نفرون دیستال میباشند.
بیمارانی که در آنها غلظت کلر در ادرار بالاست و آلکالوز دارند و مبتلا به پرفشاری خون میباشند، باید از نظر هیپرکوتیسیزم بررسی گردند. هیپرکورتیسیزم ممکن است حالت خودمختار داشته باشد (مانند آلدوسترونیسم اولیه و بیماری کوشینگ) یا ثانویه به تنگی شریان کلیه باشد. اختلالات نادرتر، عبارتاند از کمبودهای β11- هیدروکسیلاز یا افزایش ظاهری مینرالوکورتیکوئید که در آن، کاهش تبدیل (conversion) گلوکزکورتیکوئیدهایی که به مجرای جمعکننده میرسند، منجر به افزایش تحریک ENaC میشود. سندرم دیگری که با هیپرتانسیون مادرزادی و آلکالور همراه است، آلدوسترونیسم قابل تصحیح با گلوکوکوتیکوئید (GRA)، یک مضاعف شدن ژنی است؛ بدین صورت که پیشبرندهٔ (promoter) ژن β11- هیدروکسیلاز، ژن آلدوسترون سنتاز را تحریک میکند و منجر به ساخت آلدوسترون پاسخدهنده به هورمون آدرنوکورتیکوتروپیک میگردد. در هر کدام از این موارد، افزایش فعالیت ENaC منجر به تمام خصوصیات بالینی سندرم میشود.
آلکالوز همراه با فشار خون طبیعی یا پایین، هیپوکالمی و بالا بودن غلظت کلر ادرار، دو تشخیص افتراقی را مطرح میکند: سندرم بارتر و سندرم گیتلمن. در هر یک از این سندرمها، ناهنجاریهای مختلفی در بازجذب کلرید سدیمِ اختصاصی برای قطعه وجود دارد. همچنین دفع کلسیم و منیزیم در این دو سندرم با هم متفاوت است. در سندرم بارتر (جدول 6 ـ 28)، جهشهای مختلف در ژنهای تأثیرگذار بر بازجذب کلریدسدیم در شاخهٔ ضخیم صعودی، این ژنها را غیرفعال میکنند. ازجملهٔ این جهشها، ژنهای کاهشدهندهٔ فعالیت در هم انتقالدهندهٔ NKCC2، پروتئین کانال ROMK و کانال کلر واقع در غشای قاعدهای ـ جانبی و نیز جهش افزایندهٔ فعالیت در گیرندهٔ حسکنندهٔ کلسیم میباشند. هر کدام از این جهشها، منجر به اتلاف نمک، افزایش دفع کلسیم، کاهش حجم و در بسیاری موارد، کاهش فشارخون میشوند. در اثر کاهش حجم ECF، هیپرآلدوسترونیسم ثانویه ایجاد میشود که در همراهی با افزایش رسیدن سدیم به مجرای جمعکننده، منجر به اتلاف پتاسیم و افزایش دفع پروتون میگردد. در سندرم گیتلمن، جهشهای ناتوانکننده در هم انتقالدهندهٔ سدیم ـ کلر حساس به تیازید در لولهٔ پیچیدهٔ دور صورت میگیرد. مشخصات فنوتیپی سندرم گیتلمن که در تناقض با سندرم بارتر قرار دارند کاهش دفع کلسیم و هیپرکلسمی میباشند همچنان که از مهار همانتقالدهندهٔ سدیم ـ کلر انتظار میرود (فصل 26 را ملاحظه کنید). علت دفع بیش از حد منیزیم در ادرار در سندرم گیتلمن کاهش بیان کانال TRPM6 در لولهٔ پیچیدهٔ دور میباشد.
مطالب مرتبط