بتا تالاسمی یعنی چه؟ توضیح کامل بیماری خونی ارثی بتا تالاسمی و مفهوم آن
وقتی یک ژن کوچک میتواند سرنوشت زندگی یک کودک را تغییر دهد…
دختری هفتساله در یک روستای جنوبی ایران هر ماه به بیمارستان میآید. او از نوزادی با کمخونی شدید دستوپنجه نرم میکند و برای ادامه زندگی نیازمند تزریق خون منظم است. والدینش نمیدانستند حامل یک تغییر ژنتیکی پنهان هستند که وقتی با هم ازدواج کردند، احتمال ابتلای فرزندشان به بیماریای به نام «بتا تالاسمی» (Beta thalassemia) افزایش یافت. این بیماری خونی ارثی ناشی از نقص در ژن تولید زنجیره بتا در هموگلوبین است؛ همان پروتئینی که وظیفه حمل اکسیژن در گلبولهای قرمز را بر عهده دارد.
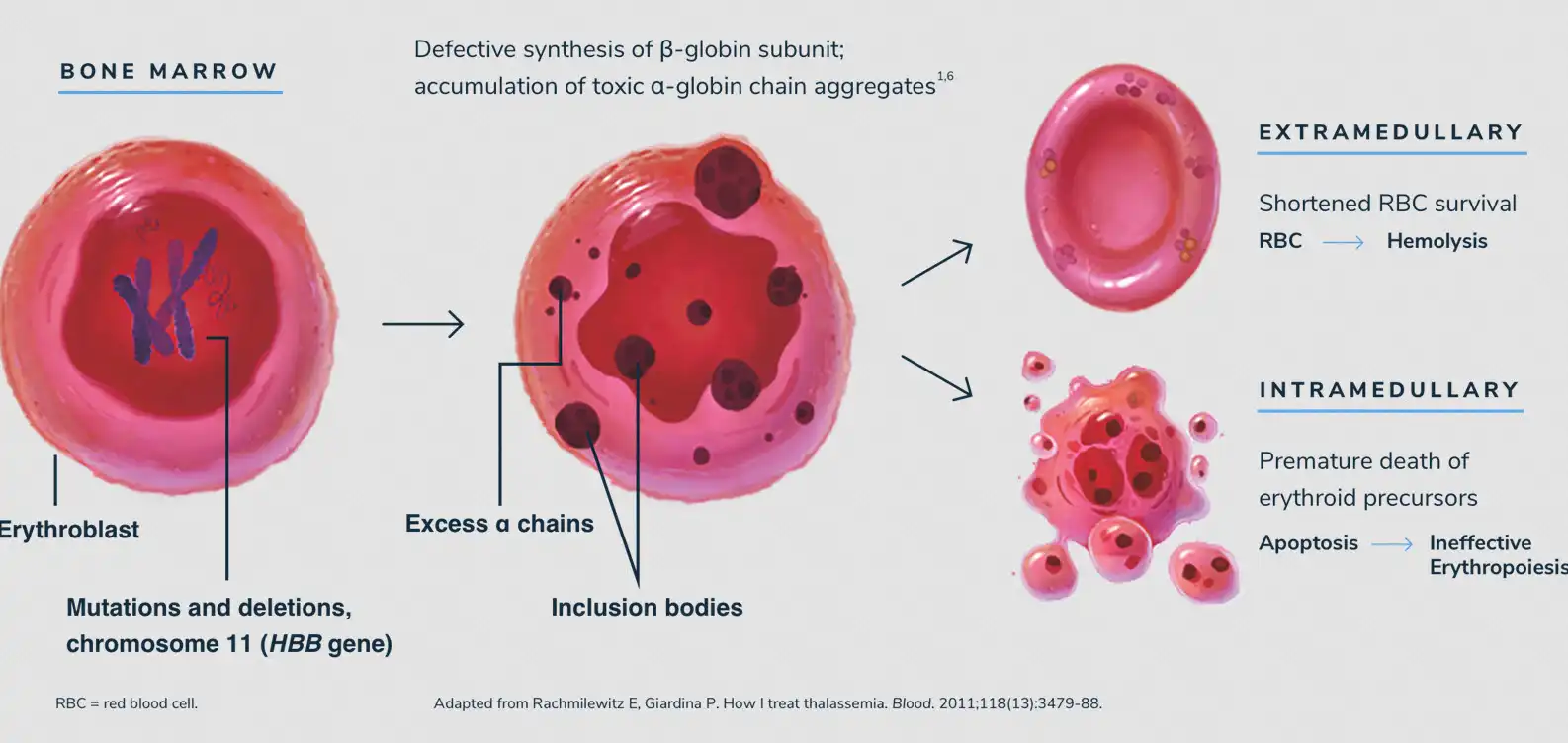
در بتا تالاسمی، هموگلوبین بهدرستی ساخته نمیشود، گلبولهای قرمز شکنندهتر میشوند و عمرشان کاهش مییابد. نتیجه آن کمخونی مزمن، ضعف، رنگپریدگی و نیاز به درمانهای طولانیمدت است. اما این بیماری فقط یک اختلال ساده خونی نیست؛ بار روانی، اقتصادی و اجتماعی بزرگی بر خانوادهها و سیستمهای درمانی میگذارد. هر تزریق خون، هر داروی شلاتکننده آهن (Iron chelator) و هر بستری در بیمارستان، بخشی از داستان پرچالش بیماران تالاسمی است.
اما سؤال مهم اینجاست: این بیماری چگونه کشف شد؟ چرا در برخی مناطق دنیا بیشتر شایع است؟ و چه راههایی برای کنترل یا پیشگیری از آن وجود دارد؟ پاسخ به این پرسشها، نهتنها جنبه علمی دارد بلکه به ما نشان میدهد که چگونه یک تغییر کوچک در DNA میتواند سرنوشت نسلها را شکل دهد.
۱- ریشه لغوی (Beta thalassemia = بتا تالاسمی)
واژه «تالاسمی» (Thalassemia) از واژه یونانی «Thalassa» به معنای دریا گرفته شده است. دلیل این نامگذاری آن بود که بیماری نخستینبار در جوامع حاشیه دریای مدیترانه مشاهده شد. بخش «بتا» (Beta) به زنجیره بتای هموگلوبین اشاره دارد که در این بیماری دچار نقص یا کاهش تولید میشود. هموگلوبین از چهار زنجیره پروتئینی تشکیل شده است: دو زنجیره آلفا (Alpha chains) و دو زنجیره بتا (Beta chains).
بنابراین اصطلاح «بتا تالاسمی» بهطور خاص به اختلالی اشاره میکند که در آن تولید زنجیرههای بتا مختل میشود. این اصطلاح در شاخههای هماتولوژی (Hematology) و ژنتیک پزشکی (Medical genetics) کاربرد دارد. به زبان ساده، ریشه لغوی اصطلاح به ما نشان میدهد که این بیماری هم منشأ جغرافیایی دارد و هم منشأ مولکولی.
۲- تاریخچه استفاده از واژه بتا تالاسمی
واژه «تالاسمی» نخستینبار در سال ۱۹۲۵ توسط دکتر توماس کولی (Thomas Cooley) برای توصیف کمخونی شدید در کودکان ایتالیایی-آمریکایی به کار رفت. او متوجه شد این کودکان از همان اوایل زندگی دچار رنگپریدگی، بزرگی طحال و مشکلات رشد میشوند. در ابتدا، بیماری «کمخونی کولی» (Cooley’s anemia) نامیده شد. با گذشت زمان و کشف نقش ژنها، واژه علمی «تالاسمی» جایگزین شد.
در دهههای بعد، با پیشرفت دانش ژنتیک مشخص شد که تالاسمی میتواند انواع مختلف داشته باشد، از جمله آلفا تالاسمی و بتا تالاسمی. اصطلاح «بتا تالاسمی» برای نخستینبار زمانی رایج شد که پژوهشگران دریافتند نقص در زنجیره بتا مسئول شکل شدیدتری از بیماری است. امروزه این اصطلاح بهطور گسترده در مقالات پزشکی، آزمایشهای ژنتیک و برنامههای غربالگری بهکار میرود.
۳- تاریخچه علمی و شناخت بالینی بیماری
در دهههای ابتدایی قرن بیستم، بتا تالاسمی بیشتر در جوامع مدیترانهای، خاورمیانه و جنوب شرق آسیا گزارش میشد. پزشکان دریافتند که الگوی انتقال بیماری خانوادگی است و مبتنی بر وراثت اتوزومال مغلوب (Autosomal recessive inheritance). با پیشرفتهای بعدی در بیوشیمی و ژنتیک، جهشهای ژنی مسئول آن شناسایی شد.
این کشف منجر به طراحی آزمایشهای تشخیصی مانند الکتروفورز هموگلوبین (Hemoglobin electrophoresis) و در ادامه آزمایشهای مولکولی برای شناسایی ژن معیوب شد. همچنین، ارتباط بیماری با مقاومت نسبی در برابر مالاریا در مناطق بومی کشف شد که توضیح میداد چرا ژن تالاسمی در برخی جمعیتها شیوع بیشتری دارد. در طول زمان، درمانها از مراقبت حمایتی ساده به سمت تزریق خون منظم و استفاده از داروهای شلاتکننده آهن برای جلوگیری از مسمومیت آهنی (Iron overload) پیش رفتند. امروزه، پیوند مغز استخوان (Bone marrow transplantation) و روشهای ژندرمانی (Gene therapy) نیز بهعنوان امیدهای جدید مطرح هستند.
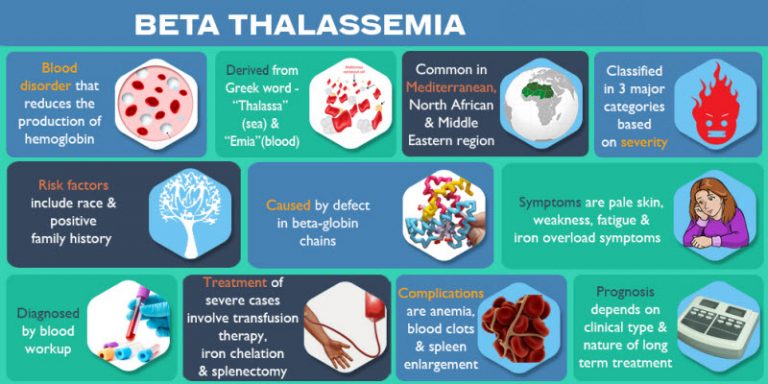
۴- انواع بالینی بتا تالاسمی و تفاوت آنها
بتا تالاسمی در سه شکل اصلی دیده میشود: مینور (Minor)، اینترمدیا (Intermedia) و ماژور (Major). در نوع مینور، فرد تنها یک نسخه معیوب از ژن را دارد و معمولاً بدون علامت یا با کمخونی خفیف است. در نوع اینترمدیا، علائم متوسطی وجود دارد و بیمار ممکن است نیاز به درمانهای پراکنده داشته باشد. نوع ماژور شدیدترین فرم بیماری است که در آن هر دو نسخه ژن بتا معیوب است و فرد از اوایل کودکی به تزریق خون منظم نیازمند میشود.
این طبقهبندی به پزشکان کمک میکند که نوع درمان و میزان مراقبت لازم را تعیین کنند. اهمیت شناخت انواع بالینی در این است که بسیاری از افراد ممکن است حامل خاموش باشند و تنها از طریق آزمایشهای ژنتیک شناسایی شوند. بنابراین، آگاهی از این تفاوتها در برنامههای پیشگیری و مشاوره ژنتیک ضروری است.
۵- علائم و تظاهرات بالینی در بیماران
علائم بتا تالاسمی عمدتاً ناشی از کمخونی شدید و تخریب مداوم گلبولهای قرمز است. بیماران دچار رنگپریدگی، ضعف، خستگی زودرس، تنگی نفس و اختلال در رشد میشوند. بزرگی طحال (Splenomegaly) و کبد (Hepatomegaly) شایع است و در موارد شدید، تغییرات استخوانی جمجمه و صورت بهدلیل فعالیت بیشازحد مغز استخوان رخ میدهد.
همچنین، رسوب آهن ناشی از تزریقهای مکرر خون میتواند به قلب، کبد و غدد درونریز آسیب برساند. مشکلات باروری، دیابت و نارسایی قلبی از پیامدهای دیررس بیماری هستند. این مجموعه علائم نشان میدهد که تالاسمی تنها یک بیماری خونی ساده نیست بلکه سندرومی پیچیده با اثرات چندسیستمی است.
۶- تشخیص بتا تالاسمی و ابزارهای آزمایشگاهی
تشخیص بیماری بر پایه ترکیبی از معاینه بالینی و تستهای آزمایشگاهی است. شمارش کامل سلولهای خون (CBC) کمخونی میکروسیتیک هیپوکروم (Microcytic hypochromic anemia) را نشان میدهد. الکتروفورز هموگلوبین افزایش HbF (هموگلوبین جنینی) و HbA2 را آشکار میسازد.
در سالهای اخیر، آزمایشهای ژنتیک مولکولی امکان شناسایی دقیق جهشهای مسئول بیماری را فراهم کردهاند. این آزمایشها نهتنها در تأیید تشخیص بلکه در برنامههای غربالگری پیش از ازدواج و پیش از تولد کاربرد دارند. تشخیص بهموقع میتواند از تولد کودکان مبتلا به تالاسمی ماژور پیشگیری کند، بهویژه در کشورهایی مانند ایران که این بیماری شیوع بالایی دارد.
۷- درمانهای رایج و چالشهای مدیریت بیماری
درمان اصلی بتا تالاسمی ماژور، تزریق خون منظم برای تأمین هموگلوبین کافی است. این کار باید همراه با داروهای شلاتکننده آهن مانند دسفرال (Deferoxamine) یا دفراسیروکس (Deferasirox) انجام شود تا از رسوب آهن در بدن جلوگیری شود. در برخی بیماران، پیوند مغز استخوان تنها درمان قطعی محسوب میشود، اگرچه هزینه و محدودیت اهداکننده مانع بزرگی است.
روشهای نوین مانند ژندرمانی در حال پیشرفت هستند و امید ایجاد کردهاند که بیماران در آینده بدون نیاز به تزریقهای مکرر زندگی کنند. با این حال، هزینههای سنگین درمان، دسترسی محدود به تجهیزات و فشار روانی بر خانوادهها همچنان چالش بزرگی باقی مانده است.
۸- نقش پیشگیری و غربالگری در کاهش شیوع بیماری
در بسیاری از کشورها، برنامههای غربالگری قبل از ازدواج بهعنوان راهی مؤثر برای پیشگیری از تولد کودکان مبتلا اجرا میشود. آزمایش خون ساده میتواند حاملان ژن تالاسمی را شناسایی کند. در صورت ازدواج دو فرد ناقل، خطر ابتلای فرزند به تالاسمی ماژور تا ۲۵ درصد افزایش مییابد.
آموزش عمومی و آگاهیرسانی نیز نقش مهمی دارد. در کشورهایی که این برنامهها بهخوبی اجرا شدهاند، نرخ تولد کودکان مبتلا بهشدت کاهش یافته است. این تجربه نشان میدهد که پیشگیری در بیماریهای ژنتیکی، گاهی مهمتر از درمان است.
۹- مقایسه بتا تالاسمی با اختلالات خونی مشابه
برای درک بهتر جایگاه بتا تالاسمی، باید آن را با بیماریهای مشابه مقایسه کرد. در «آلفا تالاسمی» (Alpha thalassemia)، نقص در زنجیرههای آلفا رخ میدهد که تظاهرات متفاوتی دارد. در «کمخونی داسی شکل» (Sickle cell anemia)، تغییر در ساختار هموگلوبین باعث تغییر شکل گلبولها میشود. «کمخونی فقر آهن» (Iron deficiency anemia) نیز با کاهش آهن بدن رخ میدهد، اما برخلاف تالاسمی علت ژنتیکی ندارد.
این مقایسه نشان میدهد که اگرچه علائم بالینی ممکن است مشابه باشند، اما مکانیسمهای زمینهای کاملاً متفاوتند. شناخت این تفاوتها برای تشخیص درست و درمان مناسب ضروری است.
خلاصه
بتا تالاسمی (Beta thalassemia) یک بیماری خونی ارثی ناشی از نقص در ژن زنجیره بتا هموگلوبین است. این بیماری در سه نوع مینور، اینترمدیا و ماژور بروز میکند و در شدیدترین حالت باعث کمخونی شدید و وابستگی به تزریق خون میشود. علائم آن شامل ضعف، رنگپریدگی، بزرگی طحال و مشکلات رشد است. تشخیص با آزمایش خون، الکتروفورز هموگلوبین و تست ژنتیک انجام میشود. درمان شامل تزریق خون، داروهای شلاتکننده آهن و در برخی موارد پیوند مغز استخوان است. پیشگیری از طریق غربالگری ژنتیک قبل از ازدواج بهترین راه کاهش شیوع بیماری محسوب میشود. بتا تالاسمی نمونهای از بیماریهای ژنتیکی است که نشان میدهد چگونه یک تغییر کوچک در DNA میتواند پیامدهای بزرگی در زندگی افراد و جامعه داشته باشد.
❓ سؤالات رایج (FAQ)
بتا تالاسمی چیست؟
یک بیماری خونی ارثی ناشی از نقص در ژن زنجیره بتا هموگلوبین که باعث کمخونی مزمن میشود.
انواع بتا تالاسمی کداماند؟
مینور (خفیف)، اینترمدیا (متوسط) و ماژور (شدید) که نیازمند تزریق خون مداوم است.
چگونه بتا تالاسمی تشخیص داده میشود؟
با آزمایش خون، الکتروفورز هموگلوبین و تست ژنتیک مولکولی.
آیا درمان قطعی برای بتا تالاسمی وجود دارد؟
پیوند مغز استخوان در برخی بیماران میتواند درمان قطعی باشد، اما محدودیتهای زیادی دارد.
چگونه میتوان از ابتلا به بتا تالاسمی پیشگیری کرد؟
با غربالگری ژنتیک پیش از ازدواج و مشاوره ژنتیک.
مطالب مرتبط