چگونه رگهای قلب دچار انسداد و گرفتگی میشود؟ بیماری عروق کرونر قلب

اپیدمیولوژی
بیماری عروق کرونر قلب (CHD) علت اصلی مرگ را در کشورهای صنعتی تشکیل میدهد. CHD علاوه بر تأثیر بر روی میزان مرگومیر بر روی ناتوانی، معلولیت و کاهش قدرت تولید نیز تأثیر میگذارد. در طی چند دههٔ گذشته به تدریج با بهبود روشهای تشخیص، پیشگیری و درمان CHD، از میزان مرگومیر ناشی از این بیماری کاسته شده است. با این وجود، تنها در ایالات متحده سالانه در حدود 1.2 میلیون مورد انفارکتوس (MI) یا حوادث قلبی کشنده روی میدهد. تقریباً نیمی از تمام مرگها در کشورهای صنعتی و 25% مرگها در کشورهای در حال توسعه به علت CHD روی میدهند. پیشبینی میشود تا سال 2020، تعداد موارد مرگومیر ناشی از CHD از تعداد مرگومیر بیماریهای عفونی پیشی گرفته و مهمترین علت مرگومیر را به خود اختصاص میدهد.
پاتوفیزیولوژی آترواسکلروز
در کشورهای صنعتی، آترواسکلروزیس اغلب در چند دههٔ اول زندگی شروع میشود. به طوریکه یکی از هر 6 نوجوان 19 ـ 13 ساله آمریکایی که به صورت اتفاقی میمیرند دارای شواهد پاتولوژیک آترواسکلروز شریانهای کرونر میباشند. تجمع لیپوپروتئینها، آسیب اندوتلیوم، و التهاب از جمله فرآیندهای متعددی هستند که در شروع و پیشرفت آترواسکلروز سهیم میباشند.
در مرحله ابتدایی آترواسکلروز، ذرات کوچک لیپوپروتئینی به اندوتلیوم رگ نفوذ نموده و در آنجا اکسید شده و در لایه انتیما انباشته میگردند. این فرآیند در محلهای آسیب اندوتلیوم تسریع میگردد که، این آسیبها ممکن است بر اثر هیپرتانسیون، هیپرکلسترولمی، مصرف سیگار، یا نیروهای برشی (shear forces) (مماسی) بیش از اندازه ایجاد شده باشند. انباشته شدن لیپید در لایه انتیما باعث بیان مولکولهای چسبیدگی (نظیر مولکول چسبیدگی داخل سلولی ـ 1، مولکول چسبیدگی سلول عروقی ـ 1، سلکتینها) موجود بر روی سطح لومینال (مجرایی) سلولهای اندوتلیال شده و بدین وسیله این امکان را به آنها میدهند که به مونوسیتهای در گردش (نظیر ماکروفاژها) متصل شوند. مونوسیتهایی که قرار است به اندوتلیوم بچسبند در پاسخ به کموکینها و سیتوکاینهای مترشحه از سلولهای اندوتلیال و سلولهای عضلانی صاف مدیای عروق در داخل لایه انتیمای عروق و در لابلای سلولهای آندوتلیال جای میگیرند. مونوسیتهای لایه انتیما با خوردن لیپوپروتئینها به مونوسیتهای انباشته از چربی یا سلولهای کفآلود (foam cells) تبدیل میشوند.
تجمع این سلولهای کفآلود، اولین شواهد قابل مشاهدهٔ آترواسکلروزیس یعنی، رگههای چربی (fattystreak) را تشکیل میدهند.
سلولهای کفآلود تکثیر یافته و با آزادسازی میانجیهای پیش التهابی سبب تداوم فرآیند التهاب موضعی و درنتیجه پیشرفت ضایعه میشوند. بعلاوه، سلولهای کفآلود آنزیمهایی ترشح میکنند که سبب تخریب اندوتلیوم میشوند.
از آنجایی که اندوتلیوم از طریق تولید مواد گشادکننده عروق نظیر پروستاسایکلین و اکسیدنیتریک (مثلاً فاکتور شلکننده مشتق از اندوتلیوم) در کنترل تون عروق و در ترومبوز دخالت دارد، آسیب سلول اندوتلیوم باعث اختلال در گشاد شدن عروق شده و به صورت موضعی یک حالت تمایل به تشکیل لخته را به وجود میآورد. پلاکتهای در گردش با چسبیدن به محل آسیب اندوتلیوم و آزادسازی فاکتورهای رشد، باعث تحریک مهاجرت و تکثیر سلولهای عضلات صاف و فیبروبلاستهای، موجود در لایه مدیا میشوند. این امر باعث تشکیل یک کلاهک فیبری بر روی هسته پر از چربی میشود.
همزمان با ادامهٔ تجمع چربی در سلولهای کفآلود، این سلولها دچار نکروز شده و چربی در مرکز پلاک باقی میماند. ماکروفاژها و ماستسلها آنزیمهای متالوپروتئیناز (نظیر کلاژناز، ژلاتیناز) را آزاد میکنند که باعث تخریب کلاژن و پروتئینهای ماترکیس خارج سلولی مجاور چربی پلاک میشوند، درحالیکه لنفوسیتهای T با ترشح سایتوکینها (نظیر اینترفرون آلفا) باعث مهار تشکیل کلاژن توسط سلولهای عضلات صاف عروق میشوند. وجود افزایش تخریب کلاژن و کاهش تشکیل آن، مجموعاً پلاک چربی را مستعد ترکخوردگی و پاره شدن مینماید. چنین پلاکهای در معرض خطر از یک هسته پر از چربی و یک کلاهک فیبری نازک تشکیل شدهاند. با ترک خوردن یا پاره شدن کلاهک فیبری نازک، کلاژن و چربی ترومبوژنیک در معرض خون در گردش قرار گرفته و درنتیجه پلاکتها به پلاک مزبور چسبیده و لخته در داخل مجرا تشکیل میگردد پلاکتهای فعال موادی را ترشح میکنند (نظیر ترومبوکسان، سروتونین) که باعث تحریک انقباض عروق و انتشار لخته میشوند. هنگامی که وسعت تجمع پلاکتها و لخته تشکیل شده به مقدار کافی برای ایجاد وقفه در جریان خون (به صورت نسبی یا کامل) میرسد، یک واقعه حاد کرونری (نظیر آنژین ناپایدار، انفارکتوس میوکارد بدون صعود قطعه [NSTEMI] ST یا انفارکتوس میوکارد همراه با صعود قطعهٔ ST [STEMI]) روی میدهد.
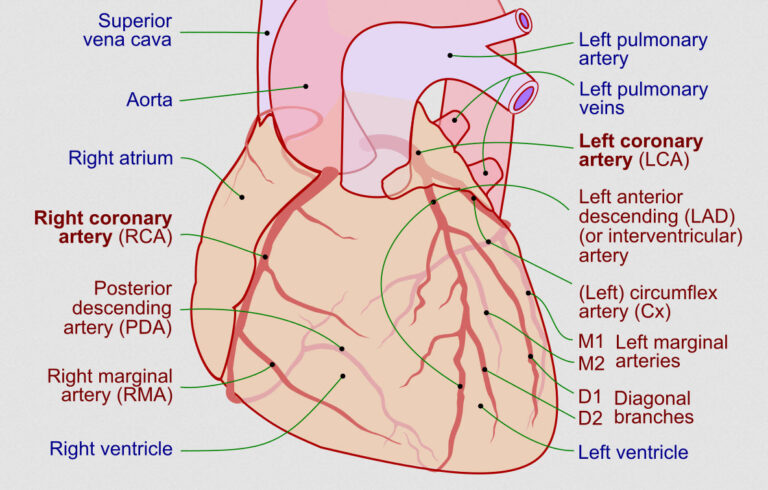
در صورتی که پلاک آترواسکلروتیک توسط یک کلاهک فیبری ضخیم پوشیده شده باشد، احتمال پارگی کمتر است، امّا ممکن است به تدریج اندازه پلاک بزرگتر شود. با بزرگتر شدن پلاک، مجرای شریان کرونری تنگ شده و جریان خونرسانی مختل میگردد (شکل 1 ـ 9). اهمیت همودینامیک پلاک به طول و شدت تنگی مجرای شریان بستگی دارد: به طور کلی، کاهش 70 درصدی در قطر مجرای شریان کرونر باعث محدودیت جریان خون در هنگام افزایش نیاز میوکارد به اکسیژن (مثلاً در طی ورزش، یا هیجانات عاطفی) شده و درنتیجه از نظر بالینی آنژین فعالیتی ایجاد میشود. یک کاهش 90 درصدی در قطر مجرا باعث ایجاد محدودیت جریان خون حتی در حالت استراحت، که نیاز میوکارد به اکسیژن طبیعی میباشد، خواهد شد. برای ضایعات با شدت بینابینی، با یک قطر تنگی مابین 40% تا 70%،؛ ارزیابی اهمیت همودینامیک تنگی به وسیلهٔ کسر جریان ذخیره (FFR «Fractional flow reserve» کسری که از حال تقسیم فشار متوسط کرونری در بعد از (دیستال) محل تنگی، که توسط یک فشارسنج بسیار کوچک مبدل فشار واقع بر روی گایدوایر آنژیوپلاستی کرونری به دست میآید، بر متوسط فشار شریانی در قبل از (پروگزیمال) محل تنگی، محاسبه میشود) یا تست ورزش میتواند در تعیین نیاز برقراری مجدد تغذیه عروقی (رواسکولاریزاسیون) کمککننده باشد.

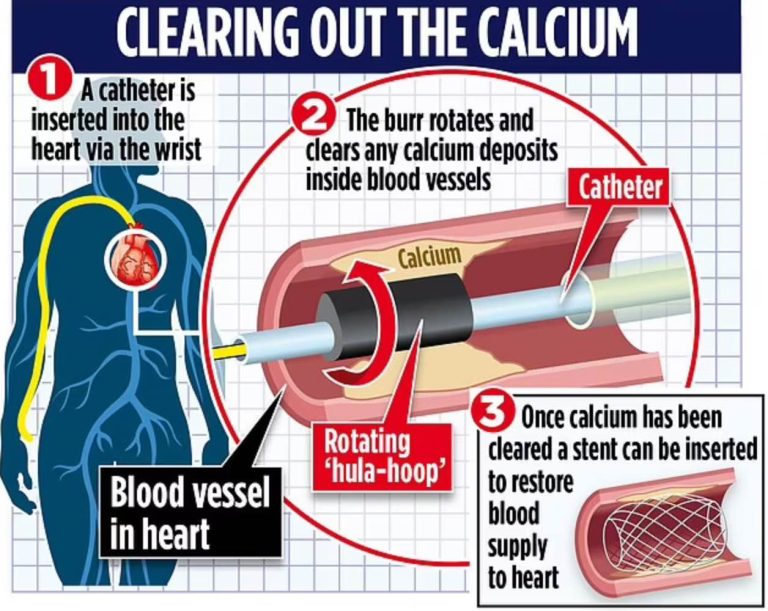
شکل 1 ـ 9. آنژیوگرام شریان کرونری راست. A. تنگی واضح قطعه میانی شریان (پیکان).B. همان شریان پس از آنژیوپلاستی موفقیتآمیز تنگی و قرار دادن استنت داخل شریان کرونر.
مطالب مرتبط