تالاسمی؛ از نقص ژنتیکی در هموگلوبین تا راهکارهای نوین مدیریت و درمان قطعی
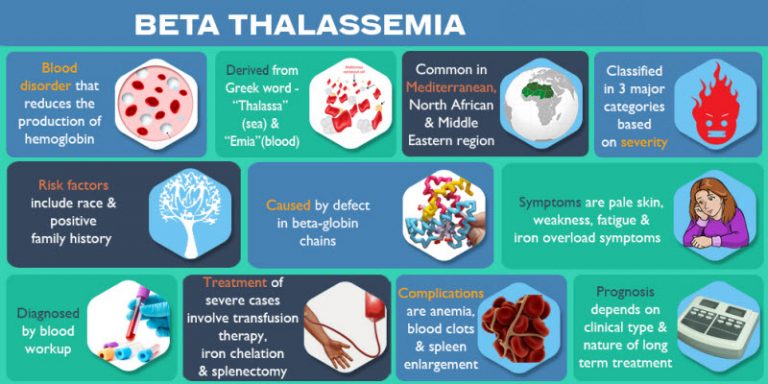
خون، این رود جاری در رگهای ما، حامل حیاتیترین عنصر بقا یعنی اکسیژن است. اما در قلب این سیستم پیچیده، گاهی یک اشتباه کوچک در کدهای ژنتیکی، سمفونی انتقال اکسیژن را مختل میکند. تالاسمی (Thalassemia) تنها یک کلمه در فرهنگ لغت پزشکی نیست؛ بلکه یک اختلال خونی ارثی است که در آن بدن توانایی ساخت مقدار کافی از پروتئین حیاتی «هموگلوبین» را از دست میدهد. هموگلوبین (Hemoglobin) همان قطاری است که اکسیژن را از ششها سوار کرده و به دورترین سلولهای بدن میرساند. وقتی این قطارها کمتر از حد استاندارد باشند یا ساختار درستی نداشته باشند، بدن در وضعیت «فقر اکسیژن» قرار میگیرد که ما آن را به نام کمخونی یا آنمی (Anemia) میشناسیم.
تالاسمی سفری است که از دوران جنینی آغاز میشود. این بیماری که ریشههای عمیقی در مناطق مدیترانهای، آسیای جنوب شرقی و آفریقا دارد، امروز به یمن پیشرفتهای علم ژنتیک، دیگر یک بنبست بیولوژیک محسوب نمیشود. از تالاسمی مینور که تنها یک ویژگی ژنتیکی ساده است تا تالاسمی ماژور که نیازمند مراقبتهای ویژه است، طیف وسیعی از چالشها و راهکارها وجود دارد. در این مقاله، ما فراتر از تعاریف کتابی، به اعماق سلولهای قرمز خون نفوذ میکنیم تا بفهمیم چگونه یک جهش ریز در DNA میتواند ساختار استخوانها، عملکرد قلب و کیفیت زندگی را تغییر دهد. با ما همراه باشید تا نقشه راه زندگی با تالاسمی را در عصر پزشکی شخصیسازی شده مرور کنیم.
“
شاید نشنیده باشید:
واژه تالاسمی از ترکیب دو کلمه یونانی «Thalassa» به معنای دریا و «Haema» به معنای خون گرفته شده است؛ چرا که پزشکان اولیه معتقد بودند این بیماری مخصوص مردمان حاشیه دریای مدیترانه است.
۱-معماری مولکولی هموگلوبین؛ وقتی زنجیرهها از هم میگسلند
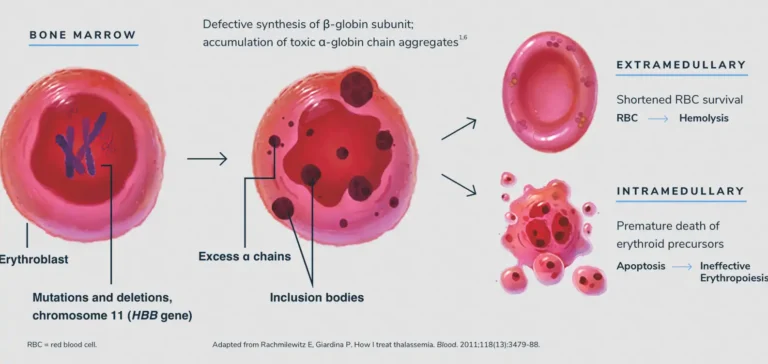
برای درک تالاسمی، ابتدا باید ساختار هموگلوبین را بشناسیم. هر مولکول هموگلوبین از دو جفت زنجیره پروتئینی ساخته شده است: دو زنجیره آلفا و دو زنجیره بتا. تالاسمی زمانی رخ میدهد که یک نقص ژنتیکی مانع از تولید کافی یکی از این زنجیرهها شود. اگر بدن نتواند زنجیره آلفا بسازد، فرد به «آلفا تالاسمی» مبتلا میشود و اگر مشکل در تولید زنجیره بتا باشد، «بتا تالاسمی» رخ میدهد. این عدم تعادل باعث میشود گلبولهای قرمز خون بسیار شکننده شوند و قبل از پایان عمر طبیعی خود (۱۲۰ روز)، تخریب گردند.
تخریب زودهنگام گلبولهای قرمز در طحال، بدن را با دو چالش بزرگ روبرو میکند: اول کمبود اکسیژن که منجر به خستگی مفرط میشود، و دوم آزاد شدن مقادیر زیادی آهن در خون. جالب است بدانید که تالاسمی در واقع یک مکانیزم دفاعی تکاملی در برابر بیماری مالاریا بوده است؛ زیرا انگل مالاریا نمیتواند در گلبولهای قرمز ناقص تالاسمی به خوبی تکثیر شود. به همین دلیل است که این بیماری در مناطقی که سابقه مالاریا داشتند، شایعتر است. اما امروزه این مزیت تاریخی، به یک چالش پزشکی مدرن تبدیل شده است که نیازمند مدیریت دقیق بیوشیمیایی است.
۲-آلفا تالاسمی؛ کالبدشکافی وراثت چهار ژنی
آلفا تالاسمی از نظر ژنتیکی پیچیدگی خاصی دارد زیرا ساخت زنجیره آلفا توسط چهار ژن (دو ژن از هر والد) کنترل میشود. شدت بیماری دقیقاً با تعداد ژنهای معیوب نسبت مستقیم دارد. اگر تنها یک ژن ناقص باشد، فرد «ناقل خاموش» است و هیچ علامتی ندارد. اما اگر دو ژن ناقص باشند، شخص دچار «صفت آلفا تالاسمی» (Alpha thalassemia trait) میشود که علائم بسیار خفیفی مانند کمخونی جزئی دارد. چالش اصلی زمانی آغاز میشود که سه ژن از چهار ژن از کار بیفتند؛ وضعیتی که به آن بیماری هموگلوبین H گفته میشود و با کمخونی متوسط تا شدید همراه است.
نادرترین و خطرناکترین حالت، نقص در هر چهار ژن است که به آن «هیدروپس فتالیس» (Hydrops fetalis) میگویند. در این وضعیت، جنین عملاً نمیتواند هیچ هموگلوبین سالمی بسازد و معمولاً قبل از تولد یا بلافاصله پس از آن از بین میرود. با این حال، پیشرفتهای علم پزشکی در دهههای اخیر امکان مداخلات داخل رحمی و پیوند سلولهای بنیادی را فراهم کرده است تا حتی این نوزادان نیز شانس حیات داشته باشند. درک این الگوهای وراثتی برای زوجهایی که قصد ازدواج یا فرزندآوری دارند، اولین سنگر دفاعی در برابر گسترش فرمهای شدید بیماری است.
۳-بتا تالاسمی؛ از دنیای مینور تا بحران ماژور
برخلاف آلفا، تولید زنجیره بتا تنها توسط دو ژن کنترل میشود. اگر یکی از این دو ژن آسیب ببیند، فرد به تالاسمی مینور (Thalassemia Minor) مبتلا است. این افراد معمولاً زندگی کاملاً نرمالی دارند و شاید تنها در آزمایش خون متوجه گلبولهای قرمز کوچکتر از حد معمول (Microcytic) شوند. مشکل اصلی زمانی رخ میدهد که هر دو ژن بتا دچار جهش شوند. در این حالت، نوزاد با تالاسمی ماژور (Thalassemia Major) یا «آنمی کولی» متولد میشود. این نوزادان در بدو تولد به دلیل وجود هموگلوبین جنینی (HbF) سالم به نظر میرسند، اما با گذشت چند ماه و جایگزینی هموگلوبین بالغ، علائم شدید ظاهر میشوند.
در بتا تالاسمی ماژور، بدن به شدت برای جبران کمخونی تلاش میکند. مغز استخوان برای تولید بیشتر خون منبسط میشود که این امر منجر به تغییر شکل استخوانهای صورت و جمجمه میگردد. همچنین، به دلیل تخریب مداوم خون، طحال و کبد بزرگ میشوند. میان این دو طیف، حالتی به نام تالاسمی اینترمدیا (Intermedia) وجود دارد که علائم آن بین مینور و ماژور متغیر است. شناسایی دقیق این زیرگروهها از طریق الکتروفورز هموگلوبین، کلید اصلی برای تعیین پروتکل درمانی است تا مشخص شود بیمار به تزریق خون نیاز دارد یا میتواند با درمانهای دارویی زندگی را سپری کند.
۴-علائم هشداردهنده؛ وقتی بدن فریاد میکشد
علائم تالاسمی مانند یک طیف نوری از نامرئی تا بسیار شدید متغیر هستند. خستگی مزمن و ضعف بدنی اولین نشانههایی هستند که ناشی از نرسیدن اکسیژن کافی به بافتهاست. پوست رنگپریده یا متمایل به زرد (یرقان ناشی از تخریب گلبولهای قرمز) نیز از علائم شایع است. در کودکان، تالاسمی خود را با تأخیر در رشد و بلوغ نشان میدهد؛ چرا که انرژی بدن به جای رشد، صرفِ تلاش بیهوده برای خونسازی میشود. همچنین ادرار تیره در این بیماران نشاندهنده دفع محصولات ناشی از تخریب هموگلوبین است.
یکی از نشانههای ظاهری خاص در موارد شدید، تغییرات استخوانی است. انبساط مغز استخوان در استخوانهای پهن باعث میشود گونهها برجسته، پیشانی پهن و فک بالایی جلوآمده به نظر برسد (Facial bone deformities). همچنین تورم شکم به دلیل بزرگ شدن طحال و کبد (Hepatosplenomegaly) در معاینات فیزیکی کاملاً مشهود است. والدین باید نسبت به این علائم در دو سال اول زندگی نوزاد بسیار حساس باشند. تشخیص زودهنگام در این مرحله میتواند از بروز تغییرات استخوانی غیرقابل بازگشت و آسیبهای قلبی ناشی از کمخونی مزمن جلوگیری کند.
۵-سایه سنگین آهن؛ پارادوکس حیات و مسمومیت
آهن برای ساخت خون ضروری است، اما در بیماران تالاسمی، همین ماده حیاتی به یک دشمن پنهان تبدیل میشود. بیماران تالاسمی ماژور به دلیل تزریقهای مکرر خون و همچنین افزایش جذب آهن از طریق دستگاه گوارش (واکنش جبرانی بدن به کمخونی)، دچار وضعیتی به نام «اضافه بار آهن» (Iron Overload) میشوند. بدن انسان مکانیسم فعالی برای دفع آهن اضافی ندارد؛ بنابراین این فلز در اعضای حیاتی مانند کبد، قلب و غدد درونریز رسوب میکند. رسوب آهن در قلب میتواند منجر به نارسایی قلبی و ریتمهای نامنظم شود که شایعترین علت مرگومیر در بیماران تالاسمی کلاسیک بوده است.
علاوه بر قلب، سیستم غدد درونریز نیز به شدت آسیبپذیر است. رسوب آهن در لوزالمعده میتواند باعث دیابت شود و آسیب به غدد جنسی منجر به تأخیر در بلوغ یا ناباروری میگردد. امروزه پزشکان با استفاده از تکنولوژیهای تصویربرداری پیشرفته مانند T2* MRI، میزان دقیق رسوب آهن در قلب و کبد را بدون نیاز به نمونهبرداری اندازه میگیرند. مدیریت این بحران نیازمند «کیلاتدرمانی» (Chelation Therapy) دقیق است؛ فرآیندی که در آن داروهایی مانند دفراسیروکس به آهن متصل شده و آن را از طریق ادرار یا مدفوع دفع میکنند. تعادل میان تزریق خون برای رفع کمخونی و دفع آهن برای جلوگیری از مسمومیت، ظریفترین بخش درمان تالاسمی است.
۶-طحالبرداری؛ جراحی در مرز ایمنی و کارایی
طحال (Spleen) نقش فیلتر خون را دارد، اما در تالاسمی، این عضو به دلیل تخریب انبوه گلبولهای قرمز دچار تورم شدید یا اسپلنومگالی (Splenomegaly) میشود. وقتی طحال بیش از حد بزرگ شود، نه تنها گلبولهای قرمز بیمار، بلکه گلبولهای تزریق شده و حتی پلاکتها را نیز زودتر از موعد تخریب میکند. در این مرحله، نیاز بیمار به تزریق خون به شدت افزایش مییابد. جراحی برداشتن طحال (Splenectomy) زمانی توصیه میشود که طحال به قدری بزرگ شود که با خطر پارگی مواجه باشد یا هزینههای تزریق خون را برای بدن غیرقابل تحمل کند.
با این حال، جراحی طحال یک شمشیر دو لبه است. طحال مرکز مهمی برای سیستم ایمنی بدن است و نبود آن، بیمار را در برابر باکتریهای کپسولدار مانند پنوموکک به شدت آسیبپذیر میکند. امروزه جراحان ترجیح میدهند این عمل را به روش لاپاروسکوپی (Laparoscopy) انجام دهند تا دوره نقاهت کوتاه شود. همچنین پروتکلهای سختگیرانهای برای واکسیناسیون قبل از عمل و مصرف آنتیبیوتیکهای پیشگیرانه تا سالها پس از جراحی وجود دارد. هدف از این جراحی، بهینهسازی طول عمر گلبولهای قرمز و کاهش دفعات مراجعه به مراکز تزریق خون است، اما نیازمند مراقبتهای بهداشتی وسواسی پس از عمل است.
۷-تشخیص پیش از تولد؛ پیشگیری به کمک تکنولوژی سلولی
یکی از بزرگترین موفقیتهای بهداشت عمومی در کنترل تالاسمی، توسعه برنامههای غربالگری پیش از ازدواج و آزمایشهای پیش از تولد (Prenatal Diagnosis) است. برای زوجهایی که هر دو ناقل (مینور) هستند، در هر بارداری ۲۵ درصد احتمال تولد نوزاد مبتلا به تالاسمی ماژور وجود دارد. امروزه با استفاده از روشهایی مانند نمونهبرداری از پرزهای جفتی (CVS) در هفته یازدهم یا آمنیوسنتز در هفته شانزدهم، میتوان وضعیت ژنتیکی جنین را با دقت بالایی تعیین کرد. این آزمایشها به والدین اجازه میدهد تا آگاهانه درباره ادامه بارداری یا آمادگی برای درمانهای بدو تولد تصمیم بگیرند.
اما فراتر از این، تکنولوژی PGD (تشخیص ژنتیکی پیش از لانهگزینی) انقلابی در این حوزه ایجاد کرده است. در این روش که با کمک لقاح آزمایشگاهی (IVF) انجام میشود، چندین جنین در آزمایشگاه تشکیل شده و تنها یک سلول از هر کدام برای بررسی ژنتیکی برداشته میشود. جراحان ژنتیک تنها جنینهایی را که فاقد نقص تالاسمی هستند انتخاب و به رحم مادر منتقل میکنند. این روش به والدین ناقل اجازه میدهد بدون ترس از سقط جنین یا تولد نوزاد بیمار، فرزندانی کاملاً سالم داشته باشند. این سطح از مهندسی بیولوژیک، عملاً زنجیره انتقال فرمهای شدید تالاسمی را در خانوادههای مستعد قطع کرده است.
“
خوب است بدانید:
ایران یکی از پیشروترین کشورها در زمینه غربالگری و پیشگیری از تالاسمی در منطقه است. به طوری که با اجرای برنامههای اجباری آزمایش خون پیش از ازدواج، نرخ تولد نوزادان مبتلا به تالاسمی ماژور در بسیاری از استانها بیش از ۸۰ درصد کاهش یافته است.
۸-ناهنجاریهای استخوانی؛ وقتی اسکلت بدن تغییر شکل میدهد
کمخونی مزمن در تالاسمی باعث میشود کلیهها هورمونی به نام اریتروپویتین را به مقدار زیاد ترشح کنند تا مغز استخوان را مجبور به خونسازی بیشتر کند. در پاسخ، مغز استخوان به شدت منبسط میشود. از آنجایی که استخوانهای کودکان نرم و در حال رشد هستند، این فشار داخلی باعث نازک شدن قشر استخوان و پهن شدن ساختار آن میشود. در جمجمه، این پدیده باعث ایجاد نمای «موهای ایستاده» (Hair-on-end) در رادیوگرافی میشود. در صورت، باعث برجستگی استخوانهای گونه و ایجاد حالتی میشود که در متون کلاسیک به آن «چهره تالاسمیک» یا سنجابگونه میگفتند.
علاوه بر تغییرات ظاهری، این انبساط باعث پوکی استخوان (Osteoporosis) و افزایش خطر شکستگیهای پاتولوژیک میشود. ستون فقرات نیز ممکن است دچار انحراف شود. امروزه با شروع به موقع تزریق خون منظم، از فعال شدن بیش از حد مغز استخوان جلوگیری میشود و بسیاری از این ناهنجاریها دیگر در نسل جدید بیماران دیده نمیشوند. با این حال، مصرف مکملهای کلسیم، ویتامین D و فعالیتهای ورزشی کنترل شده برای حفظ تراکم استخوان در این بیماران حیاتی است. این بخش از مدیریت بیماری نشان میدهد که تالاسمی نه فقط یک بیماری خونی، بلکه اختلالی است که کل سیستم اسکلتی-عضلانی بدن را تحت تأثیر قرار میدهد.
۹-پیوند سلولهای بنیادی؛ تنها پل به سوی درمان قطعی
در حالی که تزریق خون و کیلاتدرمانی تنها علائم را مدیریت میکنند، پیوند سلولهای بنیادی (Stem Cell Transplant) یا پیوند مغز استخوان، تنها راهکار موجود برای درمان قطعی تالاسمی ماژور است. در این روش، مغز استخوان بیمار که توانایی ساخت گلبولهای قرمز سالم را ندارد، با سلولهای بنیادی یک اهداکننده سازگار (معمولاً خواهر یا برادر با HLA مشابه) جایگزین میشود. اگر پیوند با موفقیت انجام شود، بدن بیمار شروع به تولید هموگلوبین نرمال میکند و نیاز به تزریق خون برای همیشه از بین میرود. بهترین زمان برای این جراحی در سنین کودکی و قبل از آسیب جدی آهن به ارگانهای حیاتی است.
با این حال، پیوند مغز استخوان یک پروسه پرخطر و گرانقیمت است. چالش اصلی، یافتن اهداکننده سازگار و مدیریت ریسک رد پیوند یا بیماری «میزبان علیه پیوند» (GVHD) است. امروزه با پیشرفت در پروتکلهای سرکوب سیستم ایمنی، شانس موفقیت در کودکان به بیش از ۸۰ تا ۹۰ درصد رسیده است. برای بیمارانی که اهداکننده فامیل ندارند، بانکهای جهانی سلولهای بنیادی و روشهای نوین جراحی با اهداکنندگان نیمهسازگار (Haploidentical) افقهای جدیدی را گشودهاند. این جراحی نه تنها یک مداخله پزشکی، بلکه آغازی دوباره برای یک زندگی بدون وابستگی به کیسههای خون است.
۱۰-تغذیه در تالاسمی؛ مدیریت هوشمندانه سفره غذایی
رژیم غذایی یک بیمار تالاسمی با یک فرد مبتلا به کمخونی معمولی کاملاً متفاوت و حتی متضاد است. برخلاف تصور عمومی، این بیماران نباید از مکملهای آهن یا مواد غذایی غنی از آهن (مانند گوشت قرمز زیاد یا جگر) استفاده کنند، زیرا مشکل آنها کمبود آهن نیست، بلکه عدم توانایی در استفاده از آن است. یکی از ترفندهای تغذیهای جالب برای این بیماران، مصرف چای همراه با غذا است؛ تانن موجود در چای باعث کاهش جذب آهن موجود در مواد غذایی گیاهی میشود. تمرکز اصلی باید بر مصرف آنتیاکسیدانها (مانند ویتامین E و C تحت نظر پزشک) برای مقابله با استرس اکسیداتیو ناشی از تجمع آهن باشد.
همچنین، به دلیل فعالیت بیش از حد مغز استخوان و پوکی استخوان احتمالی، مصرف کلسیم و ویتامین D اهمیت دوچندان پیدا میکند. لبنیات باید بخش ثابتی از رژیم غذایی باشند. از سوی دیگر، مکمل «اسید فولیک» (Folic Acid) برای این بیماران حیاتی است، زیرا بدن برای تلاش جهت ساخت گلبولهای قرمز جدید، به مقادیر زیادی از این ویتامین B نیاز دارد. مدیریت تغذیه در تالاسمی یک هنر است؛ هنری که در آن باید بدن را تقویت کرد بدون آن که انبار آهن آن را شعلهور ساخت. مشورت با یک متخصص تغذیه آشنا به اختلالات هموگلوبین، میتواند کیفیت زندگی و سطح انرژی بیمار را به طور ملموسی ارتقا دهد.
“
یک نکته کنجکاویبرانگیز:
در برخی تحقیقات مشخص شده است که مصرف غلات کامل و حبوبات به دلیل وجود فیتاتها، میتواند جذب آهن غیرهِم را تا حد زیادی کاهش دهد که این یک مزیت بزرگ برای بیماران تالاسمی در مدیریت سطح آهن بدنشان محسوب میشود.
۱۱-پیشگیری از عفونت؛ وقتی ایمنی اولویت اول است
بیماران تالاسمی، به ویژه کسانی که تحت جراحی طحالبرداری قرار گرفتهاند، در برابر عفونتها بسیار آسیبپذیرتر از افراد عادی هستند. نبود طحال یا اختلال در عملکرد آن به دلیل تجمع آهن، سد دفاعی بدن در برابر باکتریهای تهاجمی را سست میکند. کوچکترین تب در یک کودک مبتلا به تالاسمی ماژور باید به عنوان یک فوریت پزشکی تلقی شود. پیشگیری در اینجا شامل دو لایه است: واکسیناسیون کامل و منظم (از جمله واکسنهای سالانه آنفولانزا، پنوموکک و هپاتیت B) و رعایت بهداشت فردی سختگیرانه مانند شستشوی مداوم دستها و پرهیز از مصرف غذاهای نیمپز که ممکن است حاوی باکتریهای خطرناک باشند.
علاوه بر این، در فرآیند تزریق خون نیز خطر انتقال بیماریهای ویروسی همواره وجود دارد. اگرچه امروزه استانداردهای غربالگری خون بسیار بالا رفته است، اما پایش منظم از نظر هپاتیت و سایر ویروسها بخشی از پروتکلهای مادامالعمر این بیماران است. به یاد داشته باشید که آهن زیاد در بدن، محیط کشت مناسبی برای برخی باکتریها (مانند یرسینیا) فراهم میکند. بنابراین، موفقیت در درمان تالاسمی تنها در کنترل کمخونی نیست، بلکه در ایجاد یک محیط زیستی استریل و امن برای بدنی است که سربازان دفاعی کمتری در اختیار دارد.
۱۲-آمادگی برای ویزیت؛ مدیریت دادههای خونی
ویزیتهای هماتولوژی (Hematology) برای بیماران تالاسمی فراتر از یک چکآپ ساده است؛ این جلسات تنظیمکننده حیات بیمار هستند. برای بهرهوری حداکثری، همیشه لیستی از آخرین سطح «فریتین» (Ferritin) خون خود داشته باشید؛ عددی که نشاندهنده میزان ذخیره آهن بدن است. همچنین اگر علائمی مانند تپش قلب، تنگی نفس هنگام ورزش یا تغییر در رنگ پوست (تیره شدن غیرعادی) دارید، حتماً به پزشک اطلاع دهید، زیرا اینها میتوانند نشانههای اولیه درگیری قلبی یا نارسایی غدد باشند. یادداشت کردن دقیق دوز داروهای کیلاتور و هرگونه حساسیت به کیسههای خون، به پزشک کمک میکند تا پروتکل تزریق را بهینهسازی کند.
از پزشک خود درباره تکنولوژیهای نوین مانند داروهای جدید محرک بلوغ گلبول قرمز (مانند Luspatercept) سوال کنید که ممکن است نیاز به تزریق خون را کاهش دهند. همچنین اگر قصد انجام فعالیت ورزشی حرفهای یا سفر به مناطق مرتفع (که اکسیژن کمتری دارند) را دارید، حتماً مشاوره بگیرید. تالاسمی یک بیماری پویا است و مدیریت آن نیازمند همکاری نزدیک بین بیمار و تیم پزشکی شامل هماتولوژیست، متخصص قلب و پرستاران بخش خون است. پوشیدن دستبندهای هشدار پزشکی که نوع بیماری و گروه خونی شما را مشخص میکند، یک اقدام پیشگیرانه هوشمندانه برای موارد اورژانسی است.
۱۳-مهندسی ژنتیک و CRISPR؛ پایان عصر تزریق خون؟
ما در آستانه یکی از بزرگترین تحولات تاریخ پزشکی برای درمان تالاسمی هستیم. تکنولوژی اصلاح ژن یا کریسپر (CRISPR-Cas9) اکنون دیگر یک رویای علمی-تخیلی نیست. در این روش، دانشمندان سلولهای بنیادی خودِ بیمار را استخراج کرده و در آزمایشگاه، کدهای ژنتیکی آنها را به گونهای اصلاح میکنند که بدن دوباره قادر به تولید هموگلوبین سالم یا هموگلوبین جنینی (HbF) شود. سپس این سلولهای اصلاح شده به بدن بیمار بازگردانده میشوند. این روش انقلابی، نیاز به پیدا کردن اهداکننده سازگار را حذف میکند و خطر رد پیوند را به صفر میرساند، زیرا بیمار در واقع اهداکننده خودش است.
علاوه بر اصلاح ژن، روش «ژندرمانی» (Gene Therapy) با استفاده از ناقلهای ویروسی برای وارد کردن یک نسخه سالم از ژن هموگلوبین به سلولهای بیمار، نتایج درخشانی در کارآزماییهای بالینی داشته است. بسیاری از بیمارانی که در این طرحها شرکت کردهاند، اکنون ماهها یا سالهاست که بدون نیاز به حتی یک واحد تزریق خون زندگی میکنند. اگرچه این درمانها هنوز بسیار گرانقیمت و محدود به مراکز فوقپیشرفته هستند، اما نشاندهنده مسیری هستند که در آن تالاسمی ماژور از یک بیماری مزمن و وابسته به بیمارستان، به یک وضعیت قابل درمان قطعی در بدو تولد تبدیل خواهد شد.
۱۴-سلامت روان و تابآوری؛ جنبه پنهان مدیریت بیماری
زندگی با یک اختلال خونی مزمن که نیازمند مراجعات مکرر به بیمارستان است، میتواند فشار روانی سنگینی بر بیمار و خانواده او وارد کند. «خستگی از درمان» (Treatment Fatigue) پدیدهای است که در آن بیمار به دلیل تکرار مداوم تزریقها و مصرف داروها، دچار فرسودگی شده و ممکن است در رعایت پروتکلها سستی کند. حمایت روانشناختی برای این بیماران به اندازه چکآپهای قلبی اهمیت دارد. پیوستن به گروههای حمایتی و صحبت با کسانی که چالشهای مشابهی دارند، حس انزوا را کاهش داده و به افزایش تابآوری کمک میکند.
نوجوانان مبتلا به تالاسمی در سنین بلوغ با چالشهای تصویر بدنی (به دلیل تأخیر در رشد یا تغییرات استخوانی) روبرو هستند. در این مرحله، نقش خانواده در تقویت اعتمادبهنفس و تشویق به استقلال در مدیریت درمان بسیار حیاتی است. تالاسمی نباید به معنای توقف رویاها باشد؛ امروزه با درمانهای مدرن، بسیاری از این بیماران در سطوح عالی تحصیلی و شغلی فعالیت میکنند و تشکیل خانواده میدهند. سلامت روان، سوختی است که انگیزه لازم برای ادامه مسیر سخت درمان را فراهم میکند و نباید در سایه آزمایشهای خونی به فراموشی سپرده شود.
سوالات متداول (Smart FAQ)
نتیجهگیری
تالاسمی اختلالی است که در لایههای عمیق ژنتیک ریشه دارد، اما مدیریت آن در سطح زندگی روزمره رقم میخورد. از شناخت دقیق زنجیرههای آلفا و بتا تا نظارت وسواسی بر سطح آهن و استفاده از تکنولوژیهای نوین تشخیص پیش از تولد، همگی قطعات پازلی هستند که زندگی با کیفیت را برای بیماران تضمین میکنند. اگرچه تالاسمی ماژور چالشهای بزرگی را پیش روی فرد قرار میدهد، اما همگرایی مهندسی ژنتیک، جراحیهای دقیق و پروتکلهای نوین دارویی، نویدبخش آیندهای است که در آن «درمان قطعی» به جای «مدیریت علائم»، به استاندارد طلایی تبدیل خواهد شد. آگاهی، انضباط درمانی و امید، سه رکن اصلی برای عبور از سایه کمخونی و رسیدن به افقهای روشن سلامتی هستند.
تجربیات شما، راهگشای دیگران
مدیریت تالاسمی مسیری پرفراز و نشیب است. اگر شما یا عزیزانتان با این چالش روبرو هستید، از تجربیات خود در زمینه تغذیه، ورزش یا چالشهای درمان برای ما بنویسید. سوالات شما در بخش دیدگاهها توسط متخصصان ما بررسی و پاسخ داده خواهد شد تا با هم، جامعهای آگاهتر و مقاومتر بسازیم.

بیش از دو دهه در زمینه سلامت، پزشکی، روانشناسی و جنبههای فرهنگی و اجتماعی آنها مینویسد و تلاش میکند دانش را ساده اما دقیق منتقل کند.
پزشکی دانشی پویا و همواره در حال تغییر است؛ بنابراین، محتوای این نوشته جایگزین ویزیت یا تشخیص پزشک نیست.
مطالب مرتبط