پنومونیهای بینابینی ایدیوپاتیک: علایم، تشخیص و درمان

پنومونیهای بینابینی ایدیوپاتیک (IIPS)، گروهی از ILD ها (بیماریهای بینابینی ریه) با علت ناشناخته هستند. در دههٔ ۱۹۷۰، این بیماریها به طور کلی انواع مختلف IPF محسوب میشدند. با این حال، مشاهدهٔ تفاوت در تظاهرات بالینی، سیر طبیعی و پاسخ به درمان در این بیماران، منجر به تقسیمبندی مجدد آنها در گروهی از اختلالات بینابینی ایدیوپاتیک گردید. آخرین طرح طبقهبندی در بیانیه اجماع همگانی توسط انجمن قفسه سینه آمریکا و انجمن تنفسی اروپا به سال ۲۰۰۲ ابلاغ گردید. این بیماریها بهعنوان مقولههای بالینی– آسیبشناختی (کلینیکوپاتولوژیک) مجزا شناسایی شدهاند و میتوان آنها را بر اساس الگوی بافت شناختی و نیز بر اساس سیر بالینی طبقهبندی کرد.
آنهایی که سیر بالینی حاد دارند عبارتند از پنومونی بینابینی حاد (AIP)؛ سیر بالینی تحت حاد شامل پنومونی بینابینی غیر اختصاصی (NSIP)، پنومونی سازمان یابنده با منشاء ناشناخته (COP)، بیماری بینابینی ریه مرتبط با برونشیولیت تنفسی (RB-ILD)، پنومونی بینابینی لنفوئید (LIP)؛ یا یک سیر بالینی مزمن چنانکه در پنومونی بینابینی معمول usual interstitial pneumonia (UIP) دیده میشود.
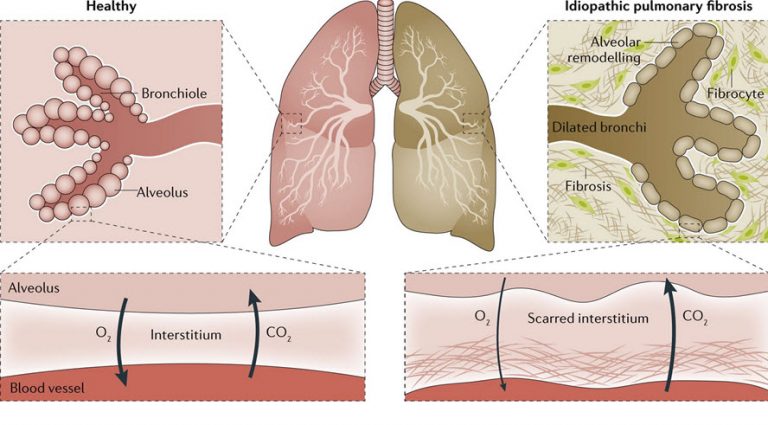
شایعترین نوع پنومونی بینابینی ایدیوپاتیک، فیبروزریوی بدون علت شناخته شده است که به آلوئولیت فیبروز دهنده با منشاء ناشناخته نیز معروف است؛ این بیماری ۸۵۰۰۰ تا ۱۰۰۰۰۰ نفر را در ایالات متحده مبتلا میکند. اگرچه در ابتدا تصور میشد که فیبروز ریوی بدون علت شناخته شده بیماری نسبتاً نادری باشد امروزه یکی از شایعترین بیماریهای بینابینی ریه تلقی میشود که شیوع آن در برخی جمعیتها به ۲۹ مورد در ۱۰۰۰۰۰ نیز میرسد؛ شیوع آن در بیماران بالای ۷۰ سال بسیار بیشتر است. در اکثر مبتلایان، بیماری به صورت تک گیر بروز میکند؛ با این حال این بیماری در افراد خانوادههای خاص نیز مشاهده شده است (فیبروز بینابینی فامیلی ریه) که بر دخالت تغییرات ژنتیکی در استعداد ابتلا به این بیماری دلالت دارد. علت بیماری ناشناخته است اما ویروسهای هرپس گاما در ریه بخش کثیری از مبتلایان به این بیماری شناسایی شده و مدل حیوانی مشابه این بیماری گزارش شده است. بسیاری از عوامل محیطی، شغلی و عفونی از جمله زیست، سیلیس، و سل میتواند باعث فیبروز ریه شوند؛ بنابراین تمایز فیبروز ریه بدون علت شناخته شده از سایر بیماریهای ریوی به علت تأثیر آن روی پیش آگهی و درمان حائز اهمیت است.
مشخصه IPF، فیبروز پیشروندهٔ ریههاست که منجربه سرفه بدون خلط و تنگی نفس میشود. این تنگی نفس، با فعالیت تشدید میشود. این تنگی نفس، تنفسی هیپوکسمیک میگردد. به طور معمول، بیمار مبتلا به IPF بین ۵۰ تا ۷۰ سال سن دارد و علائم معمولاً یک تا دو سال قبل از قطعی شدن تشخیص به وجود میآیند. معاینه فیزیکی، غالباً آشکار کننده کراکل در قاعدههای هردو ریه است و مکان غالب ایجاد اسکار را نشان میدهد. با افزایش انباشت بافت همبند، ریه سفت میشود و کاهش اتساعپذیری (کمپلیانس) ریه، گواه آن است. آزمونهای عملکرد ریوی، نشان دهنده کاهش حجمهای ریوی منطبق با فرایند محدود کننده هستند. اکسیژنرسانی ضعیف در IPF غالباً بیمار را نیازمند دریافت طولانیمدت اکسیژن مکمل میکند. رادیوگرافی قفسهٔ سینه، نشان دهندهٔ ارتشاحهایی است که بیش از همه در قاعدهها و اطراف ریه برجسته هستند. HRCT امکان مشاهدهٔ بهتر ریه را فراهم میکند و در ارزیابی گستردگی بیماری بسیار مفید است. HCRT مناطق فیبروز را مشخص میکند و اطلاعاتی در مورد ساختمانهای دیگر قفسهٔ سینه به دست میدهد. یافتههای کلاسیک HRCT در IPF، عبارتاند از ارتشاحهای دوطرفه گرهکی- مشبک با توزیع محیطی و وجود نمای لانه زنبوری و برونشکتازی کششی و عدم رویت کدورت شیشه مات (ground glass)، نفادنوپاتی و بیماری پرده جنب (شکل 3-18). در زمینه نمای بالینی بارز و یافتههای کلاسیک در HRCT، ممکن است نمونهبرداری از ریه ضروری نباشد. متاسفانه در بسیاری از بیماران برای قطعی کردن تشخیص، نیاز به انجام نمونهبرداری است.
الگوهای بافت شناختی IPF، نشان دهندهٔ مناطقی از بافت جوشگاه (اسکار) هستند که در بین آنها ساختمانهای حبابچهای طبیعی قرار دارند. یک مشخصهٔ جالب از نظر آسیبشناسی، وجود کانونهای فیبروبلاستیک است. این کانونها، محل تجمع فیبروبلاستها هستند و عقیده بر این است که محل فعالیت بیماری میباشند. این الگوی کلی سازمان یافته شدن بافتی، UIP نام دارد که میتواند در بیماریهای دیگر مانند ILD مرتبط با بافت همبند نیز دیده شود؛ بنابراین، تشخیص IPF به تابلوی بالینی، رادیوگرافیک و بافتشناسی بستگی دارد که روی هم و در غیاب یک علت واضح و تظاهرات بافت شناختی مطابق با UIP، سندرم TLD را تشکیل میدهد.
شواهد بالینی کافی در دست نیست که حاکی از بهبود بقایا کیفیت زندگی بیماران مبتلا به IPF با هر گونه درمان دارویی باشد. در نتیجه، بالینگران به طور معمول پس ازسبک سنگین کردن دقیق منافع درمان در مقابل خطرات آن بیماران را به صورت موردی مداوا میکنند.
بیماران جوانتر (زیر سال) با کدورت شیشهٔ مات قابل توجه فیبروز اندک در HRCT و کاهش حداقلی عملکرد ریه که هیچ منعی از نظر درمان نداشته باشند نامزدهای قابل قبولی برای ارائه درمان دارویی میباشند. عموماً درمان ترکیبی با استفاده از کورتیکواستروئیدها همراه با آزاتیوپرین توصیه میشود. افزودن N- استیل سیستئین به این رژیم درمانی ممکن است فواید اضافهتری دربر داشته باشد. سایر داروها از جـــــمله سیکلوفسفامید، سیکلوسپورین، کلاشیسین، پنیسیلامین و اینترفرون گاما در متوقف ساختن پیشرفت IPF نقش اندکی داشته و ممکن است مضر نیز باشند. اخیراً روشن شده که برخی مبتلایان به IPF در غیاب هرگونه علت واضخ بالینی (نارسایی قلبی، آمبولی ریوی، پنومونی) دچار وخامت حاد وضعیت تنفسی میشوند. این حملات وخامت حاد وضعیت تنفسی بدون علت شناخته شده را تشدید حاد IPF مینامند و با پیش آگهی ضعیفی همراه میباشند. یافتههای HRCT شامل کدورتهای جدید از نوع شیشهٔ مشبک یا لانه زنبوری منطبق با UIP سوار شدهاند. از لحاظ بافتشناختی شواهدی از آسیب حاد ریوی (آسیب منتشر حبابچهای) روی UIP زمینهای را میتوان یافت.
تشدید حاد IPF به طور معمول با دوزهای بالای کورتیکواستروئیدها درمان میشود. چون بقای بیماران مبتلا به IPF در لیست انتظار برای پیوند ریه کمتر از سایر بیمارانی است که به علت موارد دیگر منتظر پیوند ریه هستند بنابراین ارجاع زود هنگام برای ارزیابی جهت پیوند باید آغاز گردد. متأسفانه میزان بقای ۵ ساله بیماران مبتلا به IPF پس از پیوند ریه فقط ۴۰ تا ۵۰ درصد است.
دومین IIP شایع، NSIP است. همانطور که از نامش پیدا است، در این بیماری تابلوی بافتشناختی غیراختصاصی دیده میشود و مشخصه آن، التهاب منتشر بینابینی است. NSIP اغلب با بیماریهای دیگری مانند اختلالات بافت همبند (برای مثال، لوپوس اریتماتوی سیستمیک، آرتریتروماتویید، پلیمیوزیت) یا پنومونیت افزایش حساسیتی همراهی دارد. همراهی با این اختلالات آنقدر قوی است که در صورت اثبات بافتشناختی NSIP، باید وجود این بیماریها، فوراً بررسی شوند. همانند IPF، بیماران ممکن است دچار تنگی نفس با پیشرفت آهسته و ارتشاحهای بینابینی دو طرفه باشند. کدورت شیشه مات غالباً در HRCT دیده میشود. NSIP بهتر از IPF به داروهای سرکوبگر ایمنی پاسخ میدهد و یک دوره آزمایشی سه ماهه مصرف این داروها باید مد نظر قرار گیرد. با این حال، گاهی NSIP با فیبروز قابل ملاحظه (NSIP فیبروز دهنده) همراه است و در این بیماران در صورت امکان، باید پیوند ریه را مد نظر قرار داد.
DIP معمولاً در افراد جوان دیده میشود و با سابقه مصرف سیگار ارتباط دارد. بیماران، دچار تنگی نفس پیشرونده و ارتشاحهای دوطرفه در عکس سینه هستند. الگوی HRCT غیر اختصاصی است و ممکن است برای تشخیص، نیاز به انجام نمونهبرداری باشد. یافتههای بافتشناسی، نشان دهندهٔ تجمع ماکروفاژهای فعال شده در فضاهای حبابچهای هستند. درمان بر استفاده از داروهای سرکوبگر ایمنی و اجتناب از مواجه شدن با دخانیات تکیه دارد. DIP اغلب با بیماری بینابینی ریه ناشی از برونشیولیت تنفسی (RB-ILD) اشتباه میشود؛ اگرچه بعضی صاحبنظران این دو بیماری را یکسان میانگارند. RB-ILD تظاهر بالینی مشابهی با DIP دارد و با مصرف دخانیات مرتبط است.
AIP یک IIP غیرشایع است. هیچ غلبهٔ جنسیتی یا ارتباط با سیگار در این بیماری وجود ندارد. تفاوت تظاهر این بیماری با سایر پنومونیهای بینابینی بدون علت شناخته شده در این است که این بیماری به صورت حادتر و همراه با تنگی نفس که درعرض روزها تا هفتهها پیشرفت میکند، ظاهر میشود و بالاستثنا منجر به نارسایی تنفسی میگردد. مبتلایان اغلب دچار یک بیماری قبلی حاکی از عفونت تنفسی فوقانی با علائم جسمانی مثل درد عضلانی، درد مفاصل، تب، لرز و کسالت هستند. الگوی بافتشناختی، نشان دهنده آسیب منتشر حبابچهای است. اگرچه یک دوره مصرف داروهای سرکوبگر ایمنی توصیه میشود. این بیماری (صرفنظر از درمان یا عدم درمان) غالباً کشنده است.
COP را اغلب در گروه IIP ها قرار میدهند زیرا از نظر بالینی، مشابه آنهاست. بیماران دچار COP، تنگی نفس تحت حاد یا مزمنی دارند که ابتدا در زمان فعالیت ظاهر میشود ولی گاهی نیز یک بیماری حاد مانند عفونت ویروسی حاد تنفسی موجب آن میشود. اختلالات بافت همبند، مواد تحریکی استنشاقی و داروها (مانند متوترکسات) میتواند علت COP باشند. از نظر بافتشناسی مشخصهٔ COP، التهاب مجاری هوایی دیستال و بافت بینابینی و انسداد فضاهای هوایی دیستال توسط فیبروبلاستها و فیبروز است. این بیماری، مشابه NSIP و DIP امکان دارد به داروهای سرکوبگر ایمنی پاسخ دهد.
هنوز هم LIP توسط بسیاری به عنوان یکی از بیماریهای لنفوپرولیفراتیو ریه تلقی میشود زیرا تصور میشد که بسیاری از موارد آن منجر به لنفوم میشوند. با این حال با پیشرفتهای اخیر در ایمونوهیستوشیمی و تکنیکهای تحلیل مولکولی، به نظر میرسد فقط موارد معدودی از LIP عملاً استحاله بدخیمی پیدا میکنند. LIP یک بیماری ناشایع است که عمدتاً در زنان دیده میشود. بیماران با تنگی نفس و سرفه با شروع تدریجی و گهگاه تب، کاش وزن، درد سینه و درد مفاصل تظاهر مییابند. موارد LIP باید از لحاظ بالینی از نظر هرگونه علت یا ارتباط شناخته شدهای مثلاً با بیماریهای کلاژن -عروقی (سندرم شوگرن)، بیماریهای روماتولوژیک (آرتریت روماتوئید) و بیماریهای نقص ایمنی (ایدز) مورد بررسی قرار گیرند. از لحاظ بافتشناسی میتوان ارتشاح سلولها از جمله لنفوسیتها، پلاسماسلها و هیستیوسیتها را درون تیغههای حبابچهای مشاهده کرد. مضافاً، در LIP هیپرپلازی پنوموسیتهای نوع II و افزایش ماکروفاژهای حبابچهای قابل رویت است. اغلب فولیکولهای لنفوئید وجود دارند که معمولاً در مسیر توزیع لنفاتیک ریوی هستند. از کورتیکواستروئیدها با میزان موفقیت متغیر برای درمان LIP استفاده شده است؛ با این حال بیشتر از یکسوم بیماران به مرحله فیبروز منتشر پیشرفت میکنند. معلوم نیست که آیا درمان سیر بیماری را تحت تأثیر قرار میدهد یا اثر قابل توجهی روی فیزیولوژی ریه دارد یا نه.
مطالب مرتبط