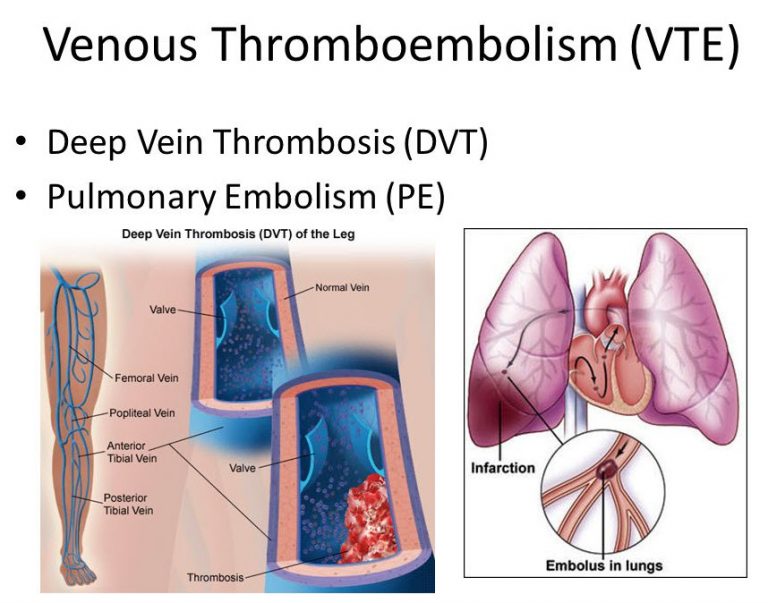
منظور از هموستاز طبیعی بدن چیست؟ بدن چگونه از ایجاد لخته بیجا پیشگیری میکند یا از خونریزی جلوگیری میکند؟
هموستاز (hemostasis) طبیعی شامل توازن فیزیولوژیک بین عوامل پیشبرنده انعقاد و عوامل ضد انعقادی بوده و سبب حفظ جریان مایع خون و انسجام ساختمانی عروق میشود. آسیب عروقی سبب شروع انعقاد با هدف تولید توپی (Plug) فیبرینی/ پلاکتی موضعی به منظور جلوگیری از خونریزی میشود. پس از آن فرآیندهایی ایجاد میشوند که سبب احتباس لخته، التیام زخم، حل لخته و باز تولید (regeneration) و بازسازی بافتی میشوند. در افراد سالم تمامی این واکنشها به طور مداوم و به گونهای متوازن روی میدهند به طوری که خونریزی بند میآید، اما در عین حال عروق خونی باز میمانند و خونرسانی کافی اعضاء را تأمین میکنند. هنگامی که به دلیل نقائص ارثی یا ناهنجاریهای اکتسابی، یک یا چند فرآیند فوق مختل میشود، هموستاز دچار اختلال میشود و ممکن است سبب عوارض خونریزی دهنده یا ترومبوآمبولی شود.
کیفیت جریان خون در تشکیلات وریدی و شریانی متفاوت است و همین امر نیازهای متفاوتی را بر سیستم انعقادی تحمیل میکند. در شریانها که فشار خون بالا است، آسیب نسبتاً جزئی عروق میتواند به سرعت منجر به خونریزی شدید شود؛ به همین دلیل پاسخ انعقادی در شریانها باید قادر به توقف سریع خونریزی باشد. پلاکتها در این پاسخ نقش اساسی دارند؛ پلاکتها ابتدا خونریزی را بند میآورند و سپس سطح فعالی به وجود میآورند که سبب شروع و تسریع تشکیل فیبرین میشود، این امر نهایتاً به هموستاز منجر میشود. از سوی دیگر، در گردش خون وریدی، سرعت جریان آهستهتر است و خونریزیهای حاصله سرعت کمتری دارند که این ویژگی سبب میشود در وریدها پلاکتها اهمیت کمتری داشته باشند؛ واکنش محوری که توازن هموستاز وریدی را کنترل میکند سرعت تولید ترومبین است. این تفاوتها در داروهای ضد انعقادی شریانی و وریدی نیز مشاهده میشوند: داروهای ضد پلاکت نظیر آسپیرین و کلوپیدوگرل برای جلوگیری از ترومبوز شریان کرونر، و مداخلات مبتنی بر آنتیترومبین نظیر هپارین و وارفارین برای پیشگیری از بروز ترومبوز ورید عمقی به کار میروند.
در این فصل به طور مختصر جزئیات فیزیولوژی هموستاز عروقی از جمله توازن طبیعی اعمال پیشبرنده انعقاد و ضد انعقاد دیواره عروق خونی، فیزیولوژی پلاکت و واکنشهای متقابل گیرنده – لیگاند (که در هموستاز نقش اساسی دارند) و فرآیندهای بسیار پیچیده و درهم تنیده آبشار انعقادی توضیح داده میشوند.
فیزیولوژی دیواره عروق
سلولهای تآندوتلیال عروق (ECs) قادر به رهبری وقایع پیش انعقادی و ضد انعقادی بر اساس شرایط هستند. این امر در بخشی مربوط به یک عملکرد سدی غیرفعل جدا کنندهٔ خون از پیش انعقادهای زیر آندوتلیال همانند کلاژن و فاکتور بافتی (TF) است. علاوه بر آن، EC های سالم به طور فعال تعادل هموستاتیک را در محیط میکروسکوپی خود از طریق محصولات ترشحی نیز تنظیم میکنند (جدول ۱-۵۲). اینها شامل پروستاسیکلین و نیتریک اکسید هستند، که هر دوی آنها سبب القای استراحت عضلهٔ صاف عروقی شده و هنگامی که در یک جهت آلبومینی آزاد شوند، فشار را کاهش میدهند. هنگامی که به درون خون ترشح میشوند، آنها سبب ایجاد آدنوزین مونوفسفات حلقوی (cAMP) پلاکت شده، و بنابراین فعالسازی و تجمع پلاکتی را مهار میکنند. سلولهای آندوتلیال همچنین آدنوزین دی فسفاتاز (ADPase) ترشح میکنند، که آدنوزین دی فسفات (ADP) خارج سلولی ترشح شده توسط پلاکتها را تجزیه میکند، و بنابراین فراخوانی پلاکتها به درون لخته یا پلاکتی در حال رشد را مهار میکند. فاکتورهای انعقادی محلول نیز سلولهای آندوتلیال هم به صورت تونیک و هم القایی تنظیم میشوند. مهارکنندهٔ مسیر فاکتور بافتی (TFPI) در فرم نوزاد خود در گردش خون، آغاز انعقاد را آهسته میکند؛ TFPI در اثر مواجهه با مقادیر اندک فاکتور Xa، آمادهٔ افزایش فعالیت است. ترومبومدولین و فعال کنندهٔ پلاسمینوژن بافتی خاموشی در ماتریکس خارج سلولی EC قرار گرفتهاند و به ترتیب آمادهٔ فعال شدن از طریق شکلگیری محلی ترومبین و فیبرین هستند، تا اعمال ضد انعقادی و فیبرینولتیپک خود را انجام دهند.
با این حال هنگامی که سلولهای آندوتلیال آسیب میبینند یا فعال میشوند، تعادل ویژگیهای انعقادی به سرعت به نفع وضعیت پیشبرندهٔ انعقاد تغییر میکند. این عمل هم از طریق خود سلولهای آندوتلیال و هم از طریق ماده زمینهای زیر آندوتلیال که بر اثر آسیب رگ نمایان میشود فعال میگردد. سلولهای اندوتلیالی فعال شده، بر سطح خود لیگاندهای چسبنده را عرضه میکنند که شامل مواد زیر هستند: سلکتینها (سلکتین E و سلکتین P)،- اینتگرین و- اینتگرین، مولکول چسبیدن پلاکت۔ سلول آندوتلیال نوع 1 (1-PECAM) و فاکتور فونویل براند (vWF)؛ (جدول ۱-۵۲ ملاحظه شود). روی سطح EC، مولتیمرهای vWF قرار گرفته و چسبیدن پلاکتی را پیش میبرند، در حالیکه اینتگرینها اتصال و مهاجرت لکوسیتها از طریق اندوتلیوم به درون بافتها را میانجیگری میکنند. ماده زمینهای زیر آندوتلیال که در تماس با جریان خون قرار گرفته است به vWF متصل میشود (شکل ۱-۵۲) و حاوی سایر اجزای چسبندهٔ پیشبرندهٔ انعقاد از جمله ترومبواسپوندین (Thrombospondin)، فیبرونکتین و کلاژن است.
| جدول 1-52. اجزای انعقادی سلولی آندوتلیال | |
| پیشبرنده انعقاد | ضدانعقاد |
| کلاژن | اتساع عروق |
| فاکتور VIII | ADPase |
| فیبرونکتین | هپاران سولفات |
| اینتگرینها | اکسید نیتریک |
| مولکول نوع ۱ چسبیدن سلول | پروستاسیکلین |
| آندوتلیال به پلاکت | ترومبومودولین |
| سلکتینهای (E و P) انقباض عروق | مهارکننده مسیر فاکتور بافتی |
| فاکتور فون ویلبراند | فعال کننده پلاسمینوژن بافتی |
این مواد، هم به عنوان لیگاند برای به دام انداختن پلاکتها عمل میکنند و هم پلاکتهای چسبیده را فعال میسازند؛ کلاژن به خصوص هم یک لیگاندو هم یک آگونیست قوی پلاکتها است که سبب میشود گرانولهای متراکم پلاکتها آزاد شوند و لیگاندهای فعالی نظیر گلیکوپروتئین GPIIb/IIIa) IIb/IIIa: ادامهٔ مطلب ملاحظه شود) در سطح پلاکتها عرضه شوند. عامل مهم دیگر پیشبرنده انعقاد که با آسیب سلول آندوتلیال نمایان میشود فاکتور بافتی tissue factor (TF) است که اساساً در سلولهای عضلانی صاف و فیبروبلاستهای زیر آندوتلیال عرضه میشود. این ماده، همچنان که بعداً اشاره خواهد شد آغازگر اصلی سیستم انعقادی محلول است که منجر به تشکیل لختهٔ فیبرینی قطعی میشود (شکل ۲-۵۲).
فیزیولوژی پلاکت
پلاکتها به عنوان یک بستر سلولی برای هموستاز عمل میکنند. گیرندههای غشای پلاکت، واسطهٔ هموستاز اولیه هستند و به پلاکتها امکان میدهند مستقیماً در نواحی آسیب، به آندوتلیوم و زیر آندوتلیوم متصل شوند. واکنش متقابل پلاکتها با گیرندههای سطحی آنها سبب پیامدهی بین غشائی میشود که منجر به فعال شدن پلاکت و پیشبرد عملیات انعقاد میشود. این کار از طریق جابجایی گیرندهها در سطح غشاء، تغییر ساختمان فضایی گیرنده، آزاد شدن محتویات گرانولها و نمایان شدن فسفولیپید غشاء صورت میگردد. سپس ورقه متشکل از پلاکتها به عنوان بستری برای مراحل متوالی آبشار انعقادی و تشکیل ترومبین عمل میکند. این سطح دو عمل زیر را انجام میدهد: ۱- برای تقویت پاسخ پیشبرندهٔ انعقاد به پلاکتها و آبشار انعقادی پس خوراند میدهد و ۲- برای ایجاد هموستاز ثانویهٔ طولانی، فیبرین تولید میکند. سرانجام، پلاکت با آزاد کردن فاکتور XIII و فاکتور پلاکتی ۴ در محیط لخته، به ترتیب سبب تحکیم لخته و محافظت آن در برابر فیبرینولیز میشود (جدول 2-52).
هموستاز پلاکتها
پلاکتهای موجود در گردش خون، سلولهای بدون هستهای هستند که قطری بین ۲ تا ۴ میکرومتر دارند و حجم آنها بین fL ۶ تا ۱۱ است. پلاکتها پس از سپری شدن دورهٔ بلوغ مگاکاریوسیتها که حدود ۴ روز است از سیتوپلاسم این سلولها منشأ میگیرند. هنگامی که پلاکتها وارد گردش خون میشوند ۷ تا ۱۰ روز دوام میآورند، ترکیبی از عامل پیری و مشارکت آنها در نگهداری طبیعی انسجام ساختمانی عروق، سبب خارج شدن پلاکتها از گردش خون میشود. در مواردی که ساختمانهای عروقی دچار گسست (مثلاً بر اثر جراحی) نشدهاند و نیز هیچ عامل استرسزایی که مصرف پلاکت را افزایش میدهد (نظیر سپسیس) وجود نداشته باشد، برای حفظ انسجام ساختمانی عروق روزانه به میزان ۷۱۰۰ پلاکت در میکرولیتر مورد نیاز است. تعداد پلاکتها در حالت طبیعی بین ۱۵۰۰۰۰ تا ۴۵۰۰۰۰ در میکرولیتر است؛ هنگامی که تعداد پلاکتها در این محدوده باشد و کار آنها طبیعی باشد، زمان سیلان (bleeding time) طبیعی (که مقیاسی از کار پلاکتها است) کمتر از ۸ دقیقه است. ترومبوسیتوپنی به تنهایی موجب طولانی شدن زمان سیلان نمیشود مگر این که تعداد پلاکتها به کمتر از ۱۰۰۰۰۰ در میکرولیتر برسد. با این حال وقتی شمار پلاکتها به کمتر از ۱۰۰۰۰۰ در مایکرولیتر برسد بر اساس زمان سیلان نمیتوان بین خونریزی ناشی از ترومبوسیتوپنی و خونریزی ناشی از غیرطبیعی بودن کار پلاکت یا اتصال آن به رگ، افتراق قائل شد. وقتی شمارش پلاکتها زیر mcL/ 100000 باشد، حتی آزمون زمان سیلان در آزمایشگاه (آنالیزور عملکرد پلاکتی [۱۰۰-]PFA) نمیتواند بین ترومبوسیتوپنی و عملکرد غیرطبیعی پلاکت افتراقی قائل شود.
چسبندگی القا شده در اثر فشار
تعامل پلاکت و دیواره رگ در جریان پرسرعت گردش خون شریانی بهتر قابل مشاهده است. واکنش متقابل عروق و جریان خون که در سمت چپ شکل ۱ – ۵۲ نشان داده شده است، لایههای موازی خون را ایجاد میکند که با سرعتهای متفاوتی در جریان هستند، لایهٔ خونی که به دیواره عروق نزدیکتر است آهستهتر از لایه خونی مرکز رگ حرکت میکند. این تفاوت سرعت، یک نیروی کششی (shear stress) ایجاد میکند که در دیواره رگ بیشتر و در مرکز آن کمتر از سایر نقاط است. بنابراین این نیروی کششی نسبت معکوس با قطر رگ دارد و میزان آن بین /Sec۵۰۰ در شریانهای بزرگ تا /Sec۵۰۰۰ در کوچکترین آرتریولها است. میزان کشش در سطح پلاکهای آترواسکلروتیک با تنگی متوسط (۵۰٪) به /See10۰۰۰-3۰۰۰ میرسد و در تنگیهایی که علایم بالینی داشته باشند میزان کششی از این هم بزرگتر میشود. جریان پر سرعت خون شریانی از طرق زیر با تشکیل لخته مقابله میکند: ۱- محدودیت زمان که اجازهٔ بروز واکنشهای پیشبرنده انعقاد را نمیدهد. ۲- جدا کردن سلولها و پروتئینهایی که اتصال سستی با جداره رگ برقرار کردهاند. اما، در صورت بروز آسیب در دیواره رگ و بروز خونریزی، پلاکتها به سرعت و قاطعانه به خللی که در یکپارچگی اندوتلیوم ایجاد شده پاسخ میدهند و به طور همزمان در برابر فشار جریان خون که می خواهد آنها را بشوید مقاومت میکنند.
یکی از نیروهایی که آمادگی پلاکت برای چسبیدن دیواره شریان را افزایش میدهد، پراکنش شعاعی (radial dispersion) است عبارت است از تمایل سلولهای بزرگتر (اریتروسیتها و لکوسیتها) برای حرکت در مرکز رگ (جایی که نیروی کششی در کمترین حد خود است) این فرآیند به نحو مؤثری پلاکتهای کوچکتر را به سمت دیواره رگ میراند و آنها را در موضع مطلوبی قرار میدهد تا بتوانند به چالشهای هموستاتیک پاسخ دهند. با استفاده از این جریان وابسته به اندازه، میتوان یافتهای به ظاهر متناقض را در مورد تزریق گویچههای قرمز خون توضیح داد و آن این است که اصلاح کمخونی شدید توسط تزریق
| جدول ۲-۵۲. ویژگیهای پیشبرنده انعقاد در پلاکتها |
| تعاملهای گیرنده – لیگاند که سبب پیشبرد چسبیدن میشوند vWF – GPIb/X/V GPIIb/IIIa فیبرینوژن و vWF – GPIIb/IIIa GPIa/IIa- کلاژن سلکتین – P-Selectin glycoprotein ligand-1 – P |
| تعاملهای لیگاند- گیرنده که سبب فعال شدن میشوند GPV- پروتئین GPVI – کلاژن |
| پروتئینهای α گرانول ترشحی لیگاندها (فیبرینوژن، فیبرونکتین، ترومبواسپوندین، ویترونکتین، vWF) آنزیمهای – آنتی پلاسمین، فاکتورهای V، VIII و XL) آنتی هپارین (فاکتور ۴ پلاکتی) |
| آگونیستهای ترشحی گرانول متراکم APD، سروتونین |
| فاکتور XIII سیتوزولی ترشحی – اجزای غشایی تشکیل ترومبوکسان A2، عرضه فسفاتیدیل سرین |
| مجموعه: CD42: GPIb/IX/V مجموعه () GPIa/IIa; CD41, GPIIb/IIIa، مجموعه گلیکوپروتئین Ia و P-Selectin glycoprotein:CD62.P.P-Selectin:CD29 vWF:CD 162, ligand 1، فاکتور فون ویلبراند |
گویچههای قرمز میتواند سبب کاهش یا توقف خونریزی شود در افرادی با اورمی شدید گردد (فصل ۵۳). این پدیده همچنین اهمیت پلاکتها در هموستاز شریانی نشان میدهد؛ افت تعداد یا کار پلاکتها ممکن است با خونریزی شریانی فاجعه باری همراه باشد. از سوی دیگر، پایین بودن میزان نیروهای کششی در گردش خون وریدی، امکان حرکت تصادفیتر سلولها و زمان بیشتر برای بروز واکنشهای انعقادی را فراهم میکند و به همین دلیل آستانه لازم برای کار و تعداد پلاکتها را کاهش میدهد.
در خونریزی شریانی که سرعت جریان خون بالا است، پلاکتها باید تقریباً بلافاصله فعال شده و به رگ آسیب دیده بچسبند. دو مولکول موجود در ناحیه زیر اندوتلیوم (فاکتور فون ویلبراند و کلاژن) در این فرایند اهمیت اساسی دارند. کنترل خونریزی در رگهایی که تحت بالاترین نیروهای کششی قرار دارند به طور قطع به وجود و کار vWF بستگی دارد. فاکتور فون ویلبراند مولکول بزرگی است که در سلولهای آندوتلیال و مگاکاریوسیتها به صورت رشتههای مولتیمر ساخته میشود؛ و vWF مولتیمر هم به طور سرشتی به جریان خون ترشح میشود و هم در اجسام ویبل پالاد (weibel-palade bodios) در EC ها ذخیره میشود. مولتیمرهای فون ویلبراند فوق بزرگ، فعالترین واحدها در اتصال پالاکت و فاکتور VIII هستند، به خصوص وقتی تحت فشار کششی قرار گرفته یا بی حرکت شوند که منجر به باز شدن چینهای آنها میشود. بزرگترین مولتیمرهای vWF که با چسبیدن به کلاژن برهنهٔ زیراندوتلیومی بی حرکت میشوند در واکنش به نیروی کششی قوی، به گیرندهٔ GPIb موجود بر سطح پلاکت متصل میشوند (شکل ۱-۵۲). این اتصال بسیار سریع روی میدهد اما میل ترکیبی موجود در آن پائین است و سبب میشود پلاکتها کنده شوند؛ اتصال مزبور موجب میگردد پلاکتها پیوند سستی با ناحیهٔ زیر آندوتلیوم برقرار کنند. در این حالت پلاکتهای چسبیده از حرکت باز میایستند و شروع به غلتیدن روی ناحیهٔ زیر آندوتلیومی میکنند. نیروی کششی زیاد همگام با سیگنالهای غشایی حاصل از واکنش متقابل GPIb- vWF منجر به از بین رفتن شکل قرص مانند طبیعی پلاکت (تغییر شکل) و تغییر شکل فضایی در گیرنده دیگر پلاکت یعنی GPIIb/IIIa میشود.
لیگاندها
اکنون مولکول فعال شدهٔ GPIIb/IIIa در محلی دور از محل اتصال GPIb/IX-V به مولتی مرهای بزرگتر vWF یا فیبرینوژن متصل میشود. میل ترکیبی در این چسبیدن ثانویه، بیش از اتصال GPIb- vWF است و اتصال مزبور سبب چسبیدن محکم پلاکتها به زیر آندوتلیوم میشود. در نیروهای کششی که تقریباً مشابه انسداد شریان است، چسبندگی پلاکت به مولتیمرهای vWF میتواند کاملاً از طریق اتصال GPIb-V-IX vWF، بدون فعالسازی پلاکت میانجیگری شود. یک عامل مهم در تنظیم فرآیند اتصال پلاکتی و فعالسازی از طریق فاکتور فون ویلبراند، پروتئاز شکافنده vWF است. این پروتئاز، یک دیسی اینتگرین و متالوپروتئیناز دارای عضو ۱۳ موتیف نوع 1 ترومبوسپوندین (ADAMTS-13) است.
ADAMTS-13 با شکافتن مولتیمرهای فوق بزرگ به قطعات کوچکتری که گرایش کلی آنها برای اتصال به پلاکتها کمتر است، فعالیت vWF را تنظیم میکند. علاوه بر فعالسازی مستقیم پلاکتها، ترومبین سبب پروتئولیز 13-ADAMTS، و بنابراین پایداری مولتیمتر بزرگ vWF شده و فراخوانی پلاکت به مناطق آسیب رگها را تقویت میکند. فقدان فعالیت پروتئاز شکافنده منجر به اتصال افسارگسیخته پلاکتها به مولتیمرهای فوق بزرگ و ترومبوز میکروواسکولار میشود (به بحث پورپورای ترومبوتیک ترومبوسیتوینیک در فصل ۵۴ مراجعه کنید).
در صورتی که میزان کشش در حد متعادلتری باشد. اتصال vWF-GPIb V-IX با اتصال پلاکت به کلاژن زیر آندوتلیومی تکمیل میشود؛ کلاژن زیر آندوتلیومی یک مادهٔ چسبنده است که با اتصال به GPla/IIa قادر است پلاکتها را متوقف کند (جدول ۲-۵۲). به این ترتیب، کلاژن و vWF زیر آندوتلیومی با همکاری همدیگر موجب شروع روند چسبیدن پلاکت میشوند و در نیروهای کششی بالاتر، Vwf نقش برجستهتری دارد.
خصوصیات منحصر به فرد کلاژن این است که با اتصال به GPla/IIa پلاکتی موجب محکم شدن پلاکتها در یک ناحیه میشود و همچنین با اتصال به GPVI پلاکتی موجب فعال شدن پلاکتها در مکان دوم میگردد و هر دو گیرندهٔ پلاکت برای فعالیت فیزیولوژیک پلاکتها حیاتی هستند. فقدان مادرزادی هر یک از گیرندههای اساسی چسبیدن پلاکت (GPIIb/IIIa، GPIbV/IX، GPVI یا GPla/IIa) سبب بروز نقائصی قابل توجه هموستاز میشود که تنها با تزریق پلاکت میتوان آنها را اصلاح نمود. زنجیرهٔ α از GPIb به طور طبیعی به عنوان یک کوفاکتور در فعالسازی پلاکتها توسط ترومبین از طریق هم گیرندهٔ GPV و هم گیرندهٔ فعال شده توسط پروتئاز (PAR) عمل میکند مشابه با نقصهای گیرنده هیا پلاکتی، کاهش در لیگاند vWF، ویژه انواع مولتیمر بزرگتر، میتواند منجربه خونریزی گردد.
پس از چسبیدن لایهای از پلاکتها به محل خونریزی، vWF متصل به GPIb بر روی فوقانیترین پلاکتها میچسبد و سبب به کارگیری پلاکتهای اضافی از جریان خون در درون توپی در حال رشد پلاکت میشود. فراخوانی پلاکتها از طریق فعالسازی سروتونین و ADP، که در جهت فعال کردن و چسبیدن پلاکتها از گردش خون به لختهٔ پلاکتی میشوند، تقویت میشود. سپس پلاکتهای چسبیده، تحت تأثیر یک سلسله فرآیندهای وابسته به هم قرار میگیرند که مجموعاً این فرآیندها را فعالسازی مینامند. فعال شدن پلاکتها پنج اثر عمده دارد: ۱- آزاد شدن موضعی لیگاندهای ضروری برای تثبیت قالب پلاکت – پلاکت، ۲- به کارگیری مداوم پلاکتهای اضافی، ۳- انقباض شریانهای کوچکتر به منظور کند کردن خونریزی، ۴- لوکالیزه کردن و تسریع تشکیل فیبرین مربوط به پلاکت و ۵- محافظت لخته در برابر فیبرینولیز.
قالب پلاکت – لیگاند – پلاکت همراه با فیبرینوژن و vWF که به عنوان پلهای عرضی (لیگاند) عمل میکنند شالوده توپی پلاکتی (platelet play) را تشکیل میدهند (شکل ۴-۵۲). هم فیبرینوژن و هم vWF در گرانولهای آلفای موجود در پلاکتهای در حال استراحت ذخیره میشوند و با فعال شدن پلاکت آزاد میشوند و هر دوی آنها میتوانند به گیرندهٔ GPIIb/IIIa هر یک از دو پلاکت متصل شوند و بدین وسیله آنها را به هم پیوند دهند. همچنان که قبلاً اشاره شد، GPIIb/IIIa پلاکتی، در حضور کلسیم، به گونهای تغییر شکل میدهد که به آن امکان میدهد به مکانی حاوی توالی اسید آمینههای آرژینین – گلیسین – آسپارتات (RGD) بر روی فیبرینوژن یا vWF متصل شود. دو مکان RGD بر روی قطبهای انتهایی هر مولکول فیبرینوژن قرار دارد، و مولتیمرهای بزرگتر vWF حاوی چندین مکان RGD هستند که همگی آنها قادرند به مولکولهای GPIIb/IIIa که دچار تغییر شکل فضایی شدهاند متصل شده یک قالب پلاکت – لیگاند – پلاکت ایجاد کنند. GPIIb/IIIa فراوانترین گلیکوپروتئین موجود بر روی سطح پلاکت است که تقریباً ۵۰۰۰۰ نسخه از آن بر سطح پلاکت در حال استراحت وجود دارد و گیرندههای GPIlb/IIIa بیشتری هم درون سیتوزول وجود دارند که پس از فعال شدن به سطح میآیند.
فعالسازی
پلاکتها تحت تأثیر آگونیستهای موضعی (کلاژن، اپینفرین و ترومبین) و نیز آزاد شدن آگونیستها از پلاکتهای موجود به محیط میکروسکوپی اطراف، وارد توپی پلاکتی میشوند و در آنجا محکم میشوند. هم کلاژن (همچنان که پیشتر اشاره شد) و هم ترومبین از طریق واکنش متقابل با گیرندههای اختصاصی پلاکت، با قدرت پلاکتها را فعال میکنند. هر چند اپینفرین به تنهایی یک آگونیست قوی پلاکت نیست، تحریک گیرنده آلفا آدرنرژیک موجود بر روی پلاکتها آنها را آماده میکند تا با اثر همافزایی (سینرژیسم) حتی در برابر آگونیستهای نسبتاً ضعیفی نظیر ADP فعال شوند. ترومبوکسان و A2 جزو ترکیبات فعال کنندهای است که مستقیماً از پلاکت آزاد میشوند، این ماده پس از شکافت اسید آراشیدونیک در مسیر سیکلواکسیژناز درون سیتوزول پلاکت تشکیل میشود و سپس در محیط لخته آزاد میگردد. ترومبوکسان و A2 هم آگونیست پلاکت و هم تنگ کننده رگ است و به سرعت به فراوردهٔ خنثی آن یعنی ترومبوکسان B2 تجزیه میشود. آسپیرین به طور برگشتناپذیری فعالیت سیکلواکسیژناز – ۱ (1-COX) را مهار میکند و در نتیجه موجب مهار تشکیل ترومبوکسان A2 در تمام طول عمر پلاکت میشود. آسپیرین با پیوند کووالان و برگشتناپذیر به ریشهٔ خاص سرین بروی 1-COX متصل میشود و با تغییرات فضایی باعث مهار جایگاه فعال واقعی آنزیم میشود که یک مولکول تیروزین متقاطع با مولکول سرین است. داروهای ضدالتهاب غیراستروئیدی (NSAIDs)، پیوند کووالان ایجاد نکرده و باعث استیلگذاری روی سرین نمیشوند. بلکه آنها پیوند برگشتپذیر و رقابتی با جایگاه فعال آنزیم که تیروزین است برقرار میکنند. بنابراین اثر ضد پلاکتی NSAID ها برخلاف آسپیرین، وابسته به سطح پلاسمایی دارو میباشد. 2-COX یک ایزوفرم القا شده درون لکوسیتهاست که التهاب و درد را میانجیگری میکند. پلاکتهای بالغ فاقد فعالیت 2-COX هستند؛ بنابراین یکی از دلایل ساخت داروهای مهارکنندهٔ فوق انتخابی 2-COX برای بیماریهای التهابی، پیشگیری از خونریزی دراثر اختلال عملکرد پلاکتی په دلیل اختلال درفعالیت 1-COX پلاکتی بود. با این وجود، به نظر میرسد BC های عروقی از فعالیت 1-COX برای تولید ترکیب ضد ترومبوژنیک پروستاسیکلین استفاده میکنند (جدول 1-52 و فصل ۵۵ را ببینید). کاهش پروستاسیکلین EC به همراه فعالیت حفظ شدهٔ بینش برندهٔ انعقاد پلاکت، ممکن است تعادل هموستاتیک یا رد جهت ایجاد لخته نگه دارد و متأسفانه، امروزه کارآزماییهای بالینی نشان دادهاند. برخی از داروهای مهارکنندهٔ فوق انتخابی 2-COX، احتمال بروز افزایش فشارخون و حوادث عروقی مانند انفارکتوس میوکارد و سکته قلبی را زیاد میکنند.
سایر آگونیستهای پلاکت بر اثر ادغام گرانولهای متراکم (dense) و گرانولهای آلفا با غشاء کانالیکولی پلاکت، به درون مایع خارج سلولی آزاد میشوند و در نتیجه محتویات گرانول به بیرون میریزد. گرانولهای متراکم حاوی سروتونین هستند که همچون ترومبوکسان A2 آگونیست پلاکت و نیز تنگ کننده رگ هستند. یکی دیگر از اجزاء گرانول متراکم، ADP است که کاملاً به شکل یک آگونیست پلاکتی عمل میکند و فاقد خواص تنگ کنندگی عروق است (جدول ۲-۵۲). اهمیت تنگی عروق ناشی از سروتونین و ترومبوکسان و A2 به طور کامل روشن نشده است. با این حال، انقباض عروق که باعث کاهش قطر رگ میشود ممکن است سبب افزایش نیروی کششی شود و در نتیجه به کارگیری پلاکتها را در ناحیه آسیب دیده را تسهیل کند. در نقایص مادرزادی گرانول متراکم (نظیر سندرم هرمانسکی ۔ پودلاک Hermansky-Pudlak syndrome) خونریزیهای شدیدی روی میدهد که این یافته اهمیت آزاد شدن گرانولهای متراکم را در حفظ هموستاز نشان میدهد. بنابراین فعال شدن پلاکتها سبب تشدید چسبندگی پلاکتها و مطلوب ساختن سطح پلاکتها برای فعالیت پیشبرندهٔ انعقاد تولید فیبرین میشود که مورد اخیر از طریق واکنش متقابل با آبشار انعقادی صورت میگردد.
انعقاد محلول
مدلهای انعقاد
مدل آبشاری انعقاد محلول (شکل ۲-۵۲ را ببینید)، که اولین بار بیش از ۴۰ سال پیش توصیف شده دو نقطهٔ آغاز را به نمایش در میآورد که به یک مسیر مشترک ختم میشوند که منجر به ایجاد ترومبین و فیبرین میگردد. این مدل اجازهٔ گامهای بلندی را در شناسایی واکنشهای پروتئولیتیک فراهم کرد که منجر به ایجاد لختهٔ فیبرینی میشوند و به خوبی با زمان آزمونهای پروترومبین (PT) و زمان نسبی تبرومبوپلاستین فعال شده (PTT) که به ترتیب تنظیم دوز وارفارین و هپارین را هدایت میکنند، در تطابق بود. با وجود آنکه در مورد برخی از سناریوهای بالینی صادق است. خونریزی در برخی از بیماریها همانند هموفیلی، پیشبینی این مدل را که هنگامی که یکی از این مسیرها غیرفعال است، فعالیت مسیر دیگر باید برای حفظ تشکیل لخته کافی باشد، نفی میکند. مدلهای تازهتر، گامهای بلندی را در روشنسازی پویایی انعقاد برداشتهاند. تنظیم پروتئینهای انعقادی توسط فعالسازی دائم و اندک فاکتور و سوار شدن کمپلکسهای آنزیمی به صورت هماهنگ است، که توسط پروتئینهای مهارکنندهای در حال گردش، در جهت منفی تنظیم میشوند. این کمپلکسهای آنزیمی شامل سرین پروتئازها، کوماکتورهای آنها و پیش مادههای زیموژن هستند. در غیاب آسیب عروقی آشکار، تشکیل کمپلکس آنزیمی و تولید ترومبین حاصل هر دو اندک و نسبتاً آهسته هستند. ضدانعقادهای در گردش برای غیرفعال کردن این کمپلکسهای پیشبرندهٔ انعقاد و جلوگیری از تشکیل لخته کافی هستند. با این حال، هنگامی که یک محرک پیشبرندهٔ انعقاد رخ میدهد، که مقادیر عمدهای از فاکتورهای فعال شده را به وجود میآورد، تشکیل این کمپلکسهای آنزیمی به سرعت تقویت میشود (بخشی از آن در اثر سردر شدن روی یک سطح غشای مناسب (فسفولیپید))، و منجر به تشکیل شدید ترومبین و در نتیجه فیبرین میگردد.
آغاز لخته
انعقاد در محیط داخل بدن به دنبال مواجههٔ خون با یک طریق شکافی از دیوارهٔ عروقی با خون تماس پیدا میکند، ایجاد میشود. مسیر داخلی یا تماسی انعقاد نقشی در اولین وقایع لختهسازی ندارد. انعقاد آغاز شده توسط TF دو فاز دارد: یک فاز آغاز و یک فاز دوم به نام انتشار (شکل ۲-۵۲ را ببینید). فاز آغاز هنگامی که TF مواجهه شده به فاکتور VIIa متصل میشود، شروع میگردد، مقادیر پیکومولار فاکتور VIIa در هر زمانی در گردش خون وجود دارد. این کمپلکسی VIIa-TF تبدیل مقادیر کوچکی از فاکتور X به Xa را کاتالیز میکند، که به نوبهای خود مقادیر نانو مولار ترومبین را به وجود میآورد. این مقادیر به نظر جزئی ترومبین که در فاز آغاز ایجاد میشود، جرقهٔ آغاز فاز انتشار را میزد، که تکمیل موفقیتآمیز آن منجر به تولید فراوان ترومبین و در نهایت، رسوب فیبرین میگردد. بیش از ۹۶٪ کل ترومبین که طی لخته ایجاد میشود، در فاز انتشار شکل میگردد.
انتشار لخته
ترومبین تولید شده طی فاز آغاز، یک فعال کنندهٔ قوی پلاکتی است، که لختهٔ در حال گسترش را از نظر غشای سطحی فعال پلاکتی و فاکتور V آزاد شده از پلاکت فراوان که فوراً توسط ترومبین به Va فعال میشود، تأمین میکند. فاکتور VIII نیز به راحتی توسط حامل خود vWF به محل خونریزی آورده شده بود، توسط ترومبین فعال میشود، این مرحله سبب آزاد شدن آن از vWF خواهد شد. سپس VIIIa با تصاویر پیکومدلار فاکتور IXa ایجاد شده توسط کمپلکسی TF-VIIIa در طی فاز آغاز واکنش میدهد، تا کمپلکس VIIla-IXa را به وجود بیاورد. تشکیل این کمپلکس بر روی سطح پلاکت پیام تغییر مسیر اولیهٔ ایجاد Xa از کمپلکس TF-VIIa (Xase خارجی) را بــه Xase داخــلی (کمپلکس VIIIa-IXa) میدهد. این تغییر بهرهٔ کینتیکی قابل توجهی دارد، زیرا کمپلکس Xase داخلی، ۵۰ برابر کارایی بیشتری از کمپلکسی Xase خارجی دارد. استعداد خونریزی مربوط به هموفیلی شاهدی از اهمیت فیزیولوژیک تولید انبوه ترومبین از طریق تغییر Xase خارجی به داخلی است. aPTT، که فاز آغاز لختهسازی شروع شده توسط یک محرک مصنوعی در محیط آزمایشگاه را اندازهگیری میکند، با کمبودهای شدید در VIII یا IX طولانی میشود، اما در هموفیلی، تولید ترومبین طی فاز انتشار، یعنی عملکردی که با aPTT اندازهگیری نمیشود، بیش از همه معیوب است.
پلاکت فعال شده، گیرندههایی را برای VIIIa و IXa بیان میکند، و اتصال این پروتئازهای فعال در ترکیب با فسفاتیدیل سرین غشا به اتصال پیش مادهٔ آنزیم، فاکتور X، را تقویت کرده و کارآیی کینتیک کمپلکسی Xase داخلی را افزایش میدهد. سوار شدن کمپلکس پروترومبیاز به طرز مشابهی برای حداکثر فعالیت به سطح غشایی فعال شدهٔ پلاکت وابسته است. همانند کمپلکس Xase، کمپلکس پروترومبیناز متصل به غشا نیز پروترومبین را نسبت به Xa آزاد عمل کننده در محلول پروترومبین، ۳۰۰, ۰۰۰ بار با میزان بالاتری فعال میکند.
Xa متصل به پلاکت، آنزیم محدود کننده در برش پروترومبین، در هر دو فاز آغاز و انتشار لختهسازی است؛ پیش مادهٔ آن، پروترومبین به GPIIb/IIIa بر روی پلاکتهای فعال و غیرفعال متصل میشود. بهرهٔ کینتیکی خالص اعطا شده در اثر اتصال پلاکت به گونهای است که سوار شدن تمامی واکنشی بروری غشای پلاکت، ۱۳ میلیون بار کارآیی کاتالیزگری پروتئاز آزاد در محلول را افزایش میدهد.
نقش سایر فاکتورهای مسیر داخلی در انعقاد چیست؟ شواهدی در حال شکلگیری است که فاکتور X فاز انتشار انعقاد را تقویت میکند. فاکتور Xa به ویژه زمانی که تغییر به xase داخلی انجام شده است، محدود کننده میباشد. با وجود آنکه مقادیر کوچک IXa توسط کمپلکسی TF-VIIa ساخته میشود. در این حالت تولید IXa توسط TFPI مهار میگردد. برای تولید Xa در مقادیر کافی جهت تأمین فاز انتشار، منبع قویتر کنیتیکی IXa مورد نیاز است. فاکتور XI زیموژن دیگری است که توسط مقادیر بسیار اندک ترومبین ساخته شده در فاز آغاز فعال میشود، اما این فعالسازی محدود به سطوح پلاکت فعال شده است. XIa متصل به پلاکت IX را بر روی سطح پلاکت فعال کرده و به این صورت به سوار شدن کمپلکس Xase داخلی کمک میکند. علاوه بر این، اتصال به سطح پلاکت، XIa را از مهارکننده آن، پروتئازنکسین ۲ محافظت میکند. بنابراین، تولید XIa بر روی پلاکت فعال شده ابزاری برای فراهم کردن IXa در مقادیر کافی جهت نگهداری حداکثر ساخت Xa از طریق کمپلکس Xase داخلی کارآمد، میباشد.
مهار انعقاد محلول
ضد انعقادهای درونساز میتوانند ترومبین شکل گرفته را مهار کنند و یا از تولید ترومبین جلوگیری کنند. مهمترین در گروه اول آنتی ترومبین (AT) است. به طور فیزیولوژیک،AT در دو برابر غلظت () بیشترین غلظت محلی ترومبین () که میتواند طی لختهسازی به دست بیاید، حضور دارد، و فعالیت AT علیه ترومبین توسط پروتئوگلیکانهای هپاران سولفات مرتبط با EC درون ساز، ۱۰۰۰ برابر تقویت میشود. غشاهای سطح پلاکت، فاکتور ۴ آزاد شده از پلاکت، از غیرفعال شدن ترومبین در لخته جلوگیریی میکنند. با این وجود، هر مقدار ترومبینی که به درون گردش خون فرار کند. بلافاصله (کمتر از ۱ دقیقه) توسط AT پلاسما مهار میشود، و در محیط میکروسکوپی FC های سالم که ۶۰۰۰۰ مولکول AT در هر سلول متصل میشود، ترومبین آزاد تقریباً به طور آنی خنثی میشود. بنابراین، تولید زودرس ترومبین به طور حیاتی به حفاظت توسط غشای پلاکت فعال شده بستگی دارد تا زمان کافی برای انتقال از فاز آغاز به انتشار وجود داشته باشد.
در میان ضدانعقادهای درونساز که ساخت ترومبین را هدف قرار میدهند، زود هنگامترین در فاز انعقاد TFPI است، که قبلاً توضیح داده شد، که فاکتور Xa و کمپلکس TF-VIIa را غیرفعال میکند.TFPI به طور ذاتی توسط EC به درون عروق میکروسکوپی آزاد میشود. در شرایط طبیعی به TFPI از طریق اتصال به گلیکوز آمینو گلیکانهای مرتبط با EC، تا حد زیادی به سطح آندوتلیال محدود است، اما میتواند توسط هپارین جابهجا شود. TFPI نوزاد تنها فعالیت مستقیم علیه Xa دارد، امایسی از برخورد با Xa، فعالیت علیه کمپلکس TF-VIIa را کسب میکند. طی فاز آغاز، Xa متصل به پلاکت از غیر فعالسازی توسط TFPI و آنتی ترومبین محافظت میشود. حفظ مقادیر کوچک Xa که طی این مرحلهٔ زودرس انعقاد تولید میشوند، برای تشکیل مقادیر نانومولار ترومبین مورد نیاز برای شروع فاز انتشار لختهسازی حیاتی است.
پروتئین فعال شده (APC) ویژگیهای ضدانعقاد، ضد التهاب و فیبرینولیتیک دارد، که آن را به تنظیم کنندهای مهم در ترومبوز و التهاب تبدیل میکند. همانند TFPI پروتئین C تنها پس اینکه انعقاد نیمه پی راه میرسد، فعال میشود. ترومبین شکل گرفته به صورت ترمبومدولین، یک پروتئوگلیکان مرتبط با سطوح آندوتلیال و سلولهای مونوسیت متصل میشود. ترومبین متصل به ترومبومدولین، توانایی خود در فعالسازی پلاکتها را از دست میدهد و به جای آن پروتئین C را فعال میکند. در سطح EC، پروتئین C نوزاد به گیرندهٔ پروتئین C سلول آندوتلیال (EPCR) متصل میشود، که آن را در موقعیتی نزدیک به ترومبین متصل شده به ترومبومدولین قرار میدهد، تا فعال شود. در واکنشی که توسط EPCR و پروتئین S تقویت میشود، APC فاکتورهای VIIIα و Va، به ترتیب اجزاء کمپلکسهای Xase و پروترومبیناز، را غیرفعال میکند و به این وسیله تقویت پیشبرندهٔ انعقاد توسط خودش را مهار میکند (شکل ۴-۵۲). همانند سایر فاکتورهای انعقاد، غشای فعال شدهٔ پلاکتی،VIIIa و Va را از غیرفعال شدن توسط APC محافظت میکند. علاوه بر اثرات APC بر ساخت ترومبین، هم چنین مهارکنندهای فعال کنندهٔ پلاسمینوژن (1-PAl) را خنثی میکند تا بازآرایی لخته را تقویت کند. APC هم چنین اثرات ضدالتهابی دارد؛ APC نوترکیب تولید فاکتور نکروز دهندهٔ تومور α را پس از چالش با اندوتوکسین کاهشی میدهد، و سوشهایی فاقد پروتئین C (هتروزیگوت) در اندوتوکسمی سیستمیک، سطوح بالاتری از سیتوکینهای پیشبرندهٔ التهاب را بروز میدهند.
کبد محل عمدهٔ ساخت تمامی فاکتورهای انعقادی است. با این وجود، در بیماری کبدی سطوح فاکتور VIII به طور کلی کاهش نمییابند، زیرا VIII توسط EC و سیستم رتیکولو آندوتلیال نیز تولید میشود. زیر گروهی از فاکتورهای انعقادی که برای ساخت وابسته به ویتامین K هستند، شامل پروترومبین (II)X IX.VII و ضد انعقادهای پروتئین C و S است، اصلاحات پس از ترجمه (از طریق یک کربوکسیلاز وابسته به ویتامین K) در دومن انتهای آمین این پروتئینها ریشههای ۱۰ – ۱۲ کربوکسی گلوتامات را اضافه میکند؛ این ریشهها برای اتصال پروتئینها و جهتیابی مناسب اتصال آنها به سطوح غشایی، حیاتی است. وارفارین اپوکسیدردوکتاز ویتامین K را متوقف میکند و بنابراین ساخت ویتامین K (از ویتامین K اپوکسید) را دوچرخهٔ ویتامین K کاهش میدهد.
آزمونهای آزمایشگاهی انعقاد
جهت اهداف آزمونهای آزمایشگاهی، آبشار انعقاد به طور مصنوعی به دو مسیر خارجی (PT) و داخلی (PTT) تقسیم میشود، که برای تشکیل یک مسیر مشترک که منجر به ساخت ترومبین و فیبرین میگردد، بدهم میپیوندند (شکل ۲-۵۲ را ببینید). در آزمایشگاه مسیر خارجی (PT) توسط اندازهگیری تداخل VIIa در گردش با TF افزوده شده از خارج (که ترومبوپلاستین نیز خوانده میشود) ارزیابی میشود. PT در حد بالایی به نقصهای فاکتورهای IX، V، VII حساس است که همهٔ آنها با خونریزی قابل توجهی همراهاند. از آنجایی که VII، II و X نیز وابسته به ویتامین K هستند، و VII کوتاهترین نیمهٔ عمر در گردش را دارد، در حال حاضر PT بهترین تست برای پایش درمان وارفارین (کومادین) میباشد. PT در اثر نقصهای IX، XI, XII و یا VIII در مسیر خارجی تحت تأثیر قرار نمیگیرد. میزان طولانی شدن PT توسط وارفارین به قدرت ترومبوپلاستین ویژه (بر اساس مقیاس بینالمللی حساسیت international sensitivity index [ISI]) و ابزار انعقاد ویژهای به کار رفته برای آزمون بستگی دارد. نسبت بینالمللی طبیعی شده international normalized ratio (INR)، این فاکتورها را جهت استانداردسازی تغییرات طولانی شدن PT که توسط وارفارین القا شده است، را در بین آزمایشگاهها محاسبه میکند. INR برای هر بیماری به این صورت محاسبه میشود: (میانگین PT / کنترل PT/ISI بیمار). INR های درمانی با وارفارین اجازهٔ کاربرد جهانی توصیههای اندیکاسیونهای ویژهای بیماری تغییر میکنند، و در فصل ۵۳ بررسی شدهاند. در مقابل وارفارین، PT نسبتاً به ضد انعقادهای درمانی L هپارین تجزیه نشده غیرحساس است.
اندازهگیری PTT بر اساس فعالسازی تماسی در محیط آزمایشگاه (مثلاً تحریک پلاسما با یک ترکیب با شارژ الکتریکی منفی همانند کائولین) انجام میگردد. PTT به نقصهای فاکتورهای تماسی (پره کالکارائین [PK]، کینینوژن با وزن مولکولی بالا [HMWK]، و فاکتور XII) و فاکتورهای انعقادی مسیر داخلی (XI, IX, VIII) و مسیرهای مشترک (پروترومبین V, X) حساس است. HMWK, PK و XII و PTT را طولانی میکنند، اما منجر به خونریزی بالینی نمیشوند، که نشان میدهد این فاکتورها به هموستاز فیزیولوژیک نامربوطاند. در مقابل، نقصیهای شدید از XI، و به ویژه IX و VIII، سبب خونریزی قابل توجه میشود. PTT همچنین به هپارین تجزیه نشده بسیار حساس است و برای پایش اثر ضدانعقاد درمانی هپارین به کار میرود. برخلاف INR برای اثر ضد انعقاد توسط وارفارین (کومارین)، گسترهٔ سطوح درمانی PTT با هپارین تجزیه نشده بسیار بزرگتر است و به سادگی قابل استانداردسازی نیست. سطوح درمانی هپارین تجزیه نشده را میتوان به وسیلهٔ آزمونهای حساس فعالیت -Xa اندازهگیری میکرد، سطوح درمانی 0.3 تا 0.7 U/mL ضد-Xa به طور کلی منجربه PTT بین 1.8 تا 5.2 برابر سطح پایداری PTT بیمار (پیش از آغاز هپارین) و یا میانگین PTT در یک جامعهٔ شاهد میگردد.
این نکته قابل ذکر است که بیشتر تستهای آزمایشگاهی مورد استفاده در انعقاد محلول، تنها کتینیک فاز آغاز را اندازهگیری میکنند. نقطهٔ پایان PTT, PT، در اولین تظاهر بر روی ژل فیبرینی هنگامی رخ میدهد که کمتر از ۵٪ کل واکنش کامل شده، و تنها سطوح اندکی از پروترومبین فعال شده است. aPTT, PT برای ردیابی ناهنجاریهای ژنتیکی در ارتباط با کمبودهای شدید فاکتور (مثلاً هموفیلی) و نیز برای هدایت درمان هپارین و وارفارین بسیار حساساند، با این حال، این آزمونها در فراهم کردن اطلاعات مربوط به تولید ترومبین طی فاز انتشار، که تعیین میکنند آیا یک لختهٔ پایدار تشکیل میشود و یا ضد انعقادهای درون زاد و تنظیم کنندههای فیبرینولیتیک از رشد اضافهٔ آن جلوگیری میکنند، ناتوان هستند.
بقا و بلوع لخته
شواهدی رو به افزایش است که تشکیل اولیهٔ یک ترومبوز، هموستاز پایدار را تثبیت میکند. پدیدههایی که طی ساخت ترومبوز آغاز میشوند و برای پایداری آن حیاتی هستند، هنگامی که لخته شکل میگردد، چه در شرایط غنی از فیبرین در ورید و چه در شرایطی غنی از پلاکت در گردش شریانی، وارد عمل میشوند.
معماری لختهٔ فیبرینی
معماری یک لختهٔ فیبرینی به شکل شگفت انگیزی متغیر است. و با وجود آنکه فاکتورهای ژنتیکی به طور قطع نقشی را در تعیین ساختار لخته برعهده دارند، دو فاکتور غالب ترومبین محلی و غلظتهای فیبرینوژن است، که واکنش آن منجربه رشتههای فیبرین میگردد. یک محیط میکروسکوپی غنی از ترومبین به طور معمول میشود و سبب میشود که در نهایت لختهٔ فیبرین به آنزیمهای لیتیک نفوذناپذیر گردد، در مقابل، در محلهای ضعیف از ترومبین، که رشتههای فیبرین کلفتتر بوده و ساختار حفرهدارتر است، لخته تمایل بیشتری به ترومبولیز دارد. به طور مشابه، غلظتهای بالای فیبرینوژن، با ترومبوزهای بزرگی همراهاند، که شبکهٔ محکمتر و سختتر آنها قابلیت تغییر شکل و مقاومت بیشتری نسبت به لیز دارد. در مقابل، غلظتهای پایین فیبرینوژن، لختههای با فشردگی کمتر و در معرض لیز تولید میکنند.
ارتباط متقاطع فیبرین توسط فاکتور XIIIA
فاکتور XIII نیز نقشی حیاتی در پایداری شکلگیری لخته برعهده دارد. XIII در پلاسما گردش میکند و نیز درون پلاکتها ذخیره میشود؛ در حقیقت، ۵۰٪ کل فعالیت پایداری فیبرین در خون درون پلاکتها حفظ شده و با فعالیت آزاد میشود. در پلاسما، XIII یک مولکول تترامر شامل دو زیر واحد α) است، که حاوی جایگاه فعال آنزیماند، و دو زیر واحد α که نیمه عمر پلاسمایی زیموژن را افزایش میدهد، اما باید برای حداکثر فعالیت آنزیم جدا شود. در مقابل، فاکتور XIII پلاکتی، دایمری است که تنها حاوی دو زیر واحد α) است. هر دو شکل زیموژن نیازمند برش ترومبین و فیبرین به عنوان یک کوفاکتور هستند، اما فعالسازی XIII پلاسمایی در سرعت بسیار کمتری پیش میرود، زیرا نیازمند جداسازی زیر واحدهای β است. XIIIa فعال شده توسط پلاسمین، به فیبرین متصل میشود و واحدهای فیبرین را به طور متقاطع متصل میکند، و بنابراین سبب کاهش نفوذپذیری و افزایش مقاومت آنها به الیز میشود. علاوه بر آن، XIIIa مهارکنندهٔ اصلی پلاسمین، α2– آنتیپلاسمین، را به طور مستقیم به فیبرین به طور متقاطع متصل میکند، و آن را در معرض خنثی شدن از هر پلاسمین پنهان شده قرار میدهد.
فیبرینولیز
سیستم فیبرینولیتیک در جهت جلوگیری از فیبرین در بستن عروق سالم عمل میکند. طی شکلگیری لخته، Xa و ترومبین، BC های سالم را تحریک میکنند تا فعال کنندهٔ پلاسمینوژن بافتی (t-PA) و فعال کنندهٔ پلاسمینوژن نوع اوروکیناز (u-PA) را آزاد کننده که هر دو قادر به برش پلاسمینوژن به پلاسمین هستند. مقادیر فراوان پلاسمینوژن حاضر در پلاسما، باعث میشود که تحت شرایط طبیعی، غلظت این آنزیمها برای تشکیل پلاسمین محدود کننده باشد. کارایی کنیتیکی t-PA حداقل چند برابر با حضور فیبرین بهبود مییابد، و بنابراین سبب نگهداری حداکثر فعالیت t-PA در محیط میکروسکوپی لخته میشود. در مقابل، به نظر میرسد u-PA نیازمند اتصال به پلاکتهای فعال برای توانایی خود در آزادمندی پلاسمین است.
میانجیهای پلاسمایی که در جهت توقف فیبرینولیز عمل میکنند، یا پلاسمین تشکیل شده را غیرفعال میکنند، (مثلاً α2 آنتیپلاسمین و احتمالاً α2 ماکروگلوبولین) یا تشکیل پلاسمین را متوقف میکنند، که مهمترین آنها
1-PAI است.1-PAI چندین برابر در پلاسما به طور اضافه وجود دارد و همچنین توسط پلاکتهای فعال شده آزاد میشود، بنابراین لختهها را از لیز زود هنگام محافظت میکند. سطوح پلاسمایی 1-PAl میتواند بسیار متغیر باشد، بخشی از آن به علت الگوی ترشح شبانه روزی آن است و بخشی به علت پلیمورفیسم در ژن 1-PAI میباشد. پلیمورفیسم 4G منطقهٔ پروموتر 1-PAI با سطوح بالاتر 1-PAI و ریسک بالاتری برای بیماری ترومبوآلبولیک مرتبط است (فصل ۵۴ را ببینید). میانجی دیگری که فیبرینولیز را در محدودهٔ لخته مهار میکند، مهارکنندهٔ فیبرینولیز فعال کنندهٔ ترومبین Thrombin activator fibrinolysie inhibitor (TAFI) است.TAFI در یک شکل غیرفعال در کبد ساخته میشود و در پلاسما، احتمالاً در ترکیبی با پلاسمینوژن، گردش میکند.TAFI ریشههای لیزین ویژهای از فیبرین را برش میدهد، که در غیر این صورت اتصال آنزیمهای فیبرینولیتیک (مثل پلاسمین) را پیش میبرند. TATI برای فعال شدن به پلاسمین و یا ترومبین نیاز دارد؛ با این وجود، فعال شلن TAFI توسط ترومبین نیازمند مقادیر بسیار فراوان و غیر معمول ترومبین آزاد است. در مقابل، ترومبومدولین مرتبط با EC، فعالسازی TAFI با القای ترومبین را ۱۲۵۰ بار تقویت میکند و بنابراین یک کوفاکتور ضروری است، که به طور غالب تنها در سطح خون – عروقی در دسترس است. علاوه بر سطح EC، ماکروفاژها نیز برای فیبرینولیز بسیار اساسی هستند. ماکروفاژها لختهٔ فیبرینی را از طریق پروتئولیز لیزوزومی توسط یک مکانیسم غیروابسته به پلاسمین تجزیه میکنند. ماکروفاژ از طریق رسپتور اینتگرین سطحی فیبرین و فیبرینوژن CDIlb/CD18 به آن متصل میشد؛ این اتصال با ورود کمپلکس به داخل لیزوزوم دنبال میشود، جایی که فیبرین و فیبرینوژن تجزیه میشوند.
ترمیم و بازسازی بافت نقاط پایان فیزیولوژیک لختهسازی هستند، که در نهایت منجر به حل شدن لختهٔ فیبرینی میشود. به علاوه،t-PA و اوروکیناز، فعال کنندههای مسیر داخلی کالیکرئین، XIIa و Xia نیز پلاسمین فعال را از پلاسمین ایجاد میکنند. اتصال پلاسمینوژن به گیرندههای سطح سلول فعال شدن آن به پلاسمین را با قرار دادن آن در مجاورت t-PA و لختهٔ فیبرینی پیش میبرد و پلاسمین را از غیرفعال شدن توسط α2 – آنتیپلاسمین در گردش (نه متصل به لخته) محافظت میکند (شکلی ۴-۵۲ را ببینید). پلاسمین در نهایت ماتکریس فیبرینی را حل میکند تا پپتیدهای محلول فیبرینی و D-dimer تولید کند و همچنین متالوپروتئینازهایی را فعال میکند که بیتر بافت آسیب دیده را تجزیه میکنند. فیبروبلاستها و لکوسیتها به درون زخم مهاجرت میکنند. در مورد لکوسیتها از طریق اتصال به سلکتین، و این سلولهای التهابی در هماهنگی با فاکتورهای رشد ترشح شده توسط لکوسیتها و پلاکتهای فعال شده (مثلاً فاکتور تغییر شکل دهندهٔ بتا) فعالیت میکنند تا ترمیم عروق و بازسازی بافت را تقویت کنند.
مطالب مرتبط