تومور بدخیم غلاف عصبی محیطی (MPNST)؛ سارکوم مهاجمی که از پوشش اعصاب ریشه میگیرد
در شبکه پیچیده ارتباطی بدن ما، اعصاب مانند کابلهای الکتریکی ظریفی هستند که توسط یک لایه محافظ به نام غلاف عصب پوشانده شدهاند. اما گاهی در اعماق این پوششهای حیاتی، سلولها راه خود را گم کرده و به تومورهای تهاجمی تبدیل میشوند که علم پزشکی آنها را «تومورهای بدخیم غلاف عصبی محیطی» (Malignant Peripheral Nerve Sheath Tumors) یا به اختصار MPNST مینامد. این نوع سرطان که در گذشته با نامهای هراسانگیزی چون نوروفیبروسارکوم شناخته میشد، یکی از نادرترین و در عین حال چالشبرانگیزترین انواع سارکوم بافت نرم است. MPNST تنها یک توده ساده نیست؛ بلکه نبردی است که در مسیر پیامرسانی بدن رخ میدهد و اغلب با دردهای مرموز و ضعفهای حرکتی ناگهانی در بازوها، پاها یا تنه خود را نشان میدهد.
آنچه این بیماری را از سایر سرطانها متمایز میکند، پیوند عمیق آن با میراث ژنتیکی و سوابق درمانی فرد است. برای افرادی که با بیماری «نوروفیبروماتوز نوع ۱» (NF1) متولد میشوند، این تومور سایهای است که باید همیشه مراقب آن باشند؛ چرا که یک تومور خوشخیم قدیمی میتواند با یک جهش ناگهانی در DNA، به یک توده بدخیم تبدیل شود. همچنین، گاهی این سرطان هدیه تلخی از درمانهای گذشته است که سالها پس از اتمام پرتودرمانی برای یک سرطان دیگر، در همان ناحیه ظاهر میشود. در این مقاله، ما به بررسی دقیق نشانههایی میپردازیم که بدن برای هشدار ارسال میکند، از تودههای در حال رشد زیر پوست تا دردهای انتشاری که نباید نادیده گرفته شوند. شناخت زودهنگام MPNST، مرز میان یک جراحی موفقیتآمیز و یک بحران جبرانناپذیر است.
“
یک نکته کنجکاویبرانگیز:
اگرچه این تومورها میتوانند در هر نقطهای از بدن ظاهر شوند، اما شایعترین محل استقرار آنها اعصاب بزرگی مانند عصب سیاتیک (Sciatic Nerve) است. به همین دلیل، گاهی علائم اولیه این سرطان با دردهای شدید کمر و پا اشتباه گرفته میشود که اهمیت معاینات دقیق عصبی را دوچندان میکند.
۱-ماهیت بیولوژیک؛ وقتی سلولهای شوان طغیان میکنند
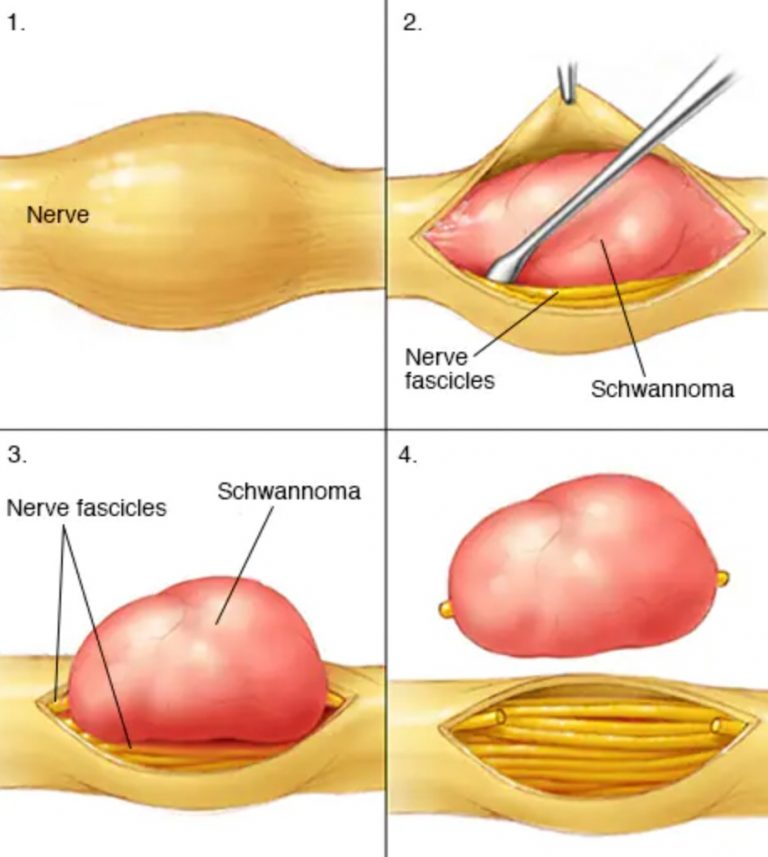
برای درک MPNST، باید به سراغ «سلولهای شوان» (Schwann cells) برویم؛ معماران اصلی غلاف میلین که اطراف اعصاب محیطی را میپوشانند. این سرطان زمانی آغاز میشود که کدهای دستوری در هسته این سلولها دچار خطاهای ویرایشی یا همان جهش (Mutation) میشوند. این جهشها باعث میشوند سلولها به جای توقف رشد، به صورت انفجاری تکثیر شوند. بر خلاف تومورهای خوشخیم که بافت اطراف را فقط کنار میزنند، تومورهای بدخیم غلاف عصبی به بافتهای مجاور یورش برده و تمایل شدیدی به گسترش (Metastasis) به ریهها و استخوانها دارند.
نکته فنی و حائز اهمیت این است که این تومورها اغلب از تغییر شکل یک «نوروفیبروم پلکسیفرم» (Plexiform Neurofibroma) پیشین ایجاد میشوند. این تغییر شکل، یک فرآیند تدریجی اما مرگبار است که در آن سلولهای خوشخیم، ویژگیهای تهاجمی پیدا میکنند. در واقع، MPNST را میتوان یکی از تهاجمیترین زیرمجموعههای سارکومهای بافت نرم دانست که به دلیل نفوذ در مسیر اعصاب، جراحی و جداسازی کامل آن بدون آسیب به عملکردهای حرکتی، به یک هنر تخصصی در جراحی مغز و اعصاب و ارتوپدی تبدیل شده است.
۲-نشانهشناسی هشداردهنده؛ فراتر از یک توده ساده
علائم MPNST اغلب فریبنده هستند و ممکن است با آسیبهای ورزشی یا دردهای عضلانی اشتباه گرفته شوند. اولین و شایعترین نشانه، وجود یک توده در حال رشد (Growing mass) در زیر پوست است که معمولاً سفت و غیرمتحرک احساس میشود. اما وجه تمایز اصلی این سرطان، دردهای انتشاری و عصبی است. از آنجا که تومور مستقیماً عصب را درگیر کرده است، بیمار ممکن است احساس تیر کشیدن، سوزش یا دردهای الکتریکی در طول یک اندام داشته باشد.
ضعف حرکتی (Motor weakness) یکی دیگر از زنگهای خطر جدی است. برای مثال، اگر تومور در عصب بازو قرار داشته باشد، فرد ممکن است به تدریج قدرت مشت کردن دست یا بالا بردن شانه را از دست بدهد. بیحسی یا احساس «گزگز» مداوم که با تغییر وضعیت بهبود نمییابد، نشاندهنده تحت فشار قرار گرفتن فیبرهای حسی عصب توسط توده بدخیم است. در موارد پیشرفته، تومور میتواند با فشار بر ریشههای عصبی در نزدیکی ستون فقرات، باعث بروز اختلال در کنترل ادرار یا حرکت کل یک اندام شود.
۳-ریشهها و محرکها؛ از میراث NF1 تا سایه پرتوها
شناختهشدهترین عامل خطر برای این سرطان، بیماری ژنتیکی «نوروفیبروماتوز نوع ۱» (Neurofibromatosis type 1) است. حدود نیمی از موارد MPNST در افرادی رخ میدهد که با این جهش ژنتیکی متولد شدهاند. برای این بیماران، خطر ابتلا به این سارکوم در طول زندگی حدود ۸ تا ۱۳ درصد تخمین زده میشود که بسیار بالاتر از جمعیت عادی است. در این افراد، تومورها تمایل دارند در سنین پایینتر (دهه ۲۰ و ۳۰ زندگی) ظاهر شوند، در حالی که در افراد عادی، سن بروز اغلب بین ۴۰ تا ۵۰ سالگی است.
عامل محرک مهم دیگر، «رادیوتراپی ثانویه» است. پارادوکس تلخ پزشکی اینجاست که پرتوهایی که زمانی برای نجات جان بیمار و از بین بردن یک سرطان دیگر (مانند سرطان لنفوم یا سینه) استفاده شدهاند، میتوانند ۱۰ تا ۲۰ سال بعد باعث ایجاد جهش در غلاف اعصاب ناحیه تحت درمان شوند. این تومورهای ناشی از اشعه، معمولاً رفتاری تهاجمیتر دارند و درمان آنها به دلیل آسیبهای قبلی بافت به وسیله پرتو، پیچیدهتر است. شناسایی این سوابق در شرححال بیمار، کلید اصلی برای شک به MPNST در مراحل اولیه است.
۴-چالشهای تشخیصی؛ ردپای سرطان در اعماق بافت
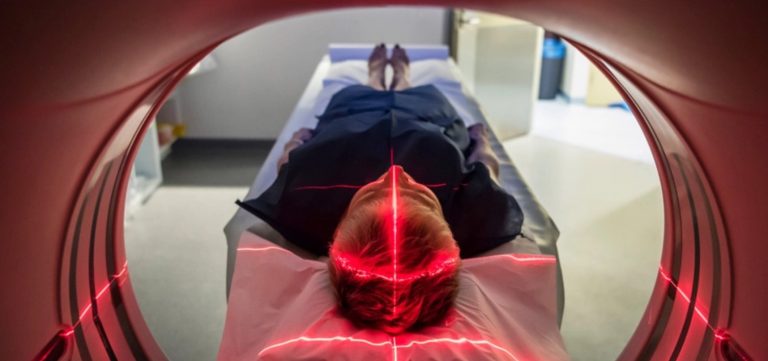
تشخیص MPNST به دلیل کمیاب بودن آن، نیازمند یک رویکرد وسواسگونه است. معاینه عصبی (Neurological exam) توسط متخصص، اولین سد دفاعی است که در آن قدرت عضلانی، بازتابها (Reflexes) و حسهای محیطی به دقت ارزیابی میشوند. اما برای دیدن آنچه در زیر پوست میگذرد، تصویربرداریهای پیشرفته الزامی است. «امآرآی» (MRI) با کنتراست بالا، استاندارد طلایی برای تعیین وسعت درگیری عصب و تفکیک تومور از بافتهای سالم اطراف است.
یک ابزار نوین و بسیار حیاتی در تشخیص این بیماری، «پت اسکن» (PET/CT) است. از آنجا که MPNSTها فعالیت متابولیک بسیار بالایی دارند، این تست میتواند به خوبی تودههای بدخیم را از نوروفیبرومهای خوشخیم متمایز کند؛ تودههایی که ممکن است در ظاهر مشابه باشند اما در پت اسکن، تومور بدخیم مانند یک نقطه نورانی پرانرژی ظاهر میشود. با این حال، حرف آخر را همیشه «بیوپسی» (Biopsy) یا نمونهبرداری بافت میزند. پزشکان باید نمونه را با دقت از ناحیهای بردارند که بیشترین فعالیت را دارد تا پاتولوژیست بتواند با بررسی میکروسکوپی، حکم قطعی بدخیمی را صادر کند.
۵-استراتژی جراحی؛ نبرد برای حذف تومور و حفظ عملکرد
ستون اصلی درمان MPNST، جراحی تهاجمی و دقیق است. هدف جراح، برداشتن کل تومور به همراه یک «حاشیه امن» (Surgical Margin) از بافت سالم اطراف است. وجود این حاشیه بسیار حیاتی است؛ زیرا اگر حتی چند سلول میکروسکوپی در لبههای جراحی باقی بماند، احتمال بازگشت (Recurrence) تومور به شدت افزایش مییابد. در تومورهایی که اعصاب اصلی دست یا پا را درگیر کردهاند، جراح با یک چالش اخلاقی و فنی بزرگ روبروست: قطع کامل عصب برای نابودی سرطان در مقابل حفظ عصب برای جلوگیری از فلج شدن اندام.
امروزه با پیشرفت تکنیکهای «جراحی حفظ اندام» (Limb-salvage surgery)، تلاش میشود تا حد امکان از قطع عضو (Amputation) جلوگیری شود. جراحان ممکن است از پیوندهای عصبی یا انتقال عضله برای بازگرداندن بخشی از عملکرد از دست رفته استفاده کنند. با این حال، در موارد بسیار تهاجمی که تومور به استخوانها یا عروق اصلی خون نفوذ کرده باشد، قطع عضو ممکن است تنها راه برای نجات جان بیمار و جلوگیری از گسترش سرطان به ریهها باشد. تصمیمگیری در این مورد معمولاً در جلسات مشترک (Tumor Board) با حضور جراحان عروق، اعصاب و ارانکولوژیستها صورت میگیرد.
۶-رادیوتراپی و شیمیدرمانی؛ سلاحهای مکمل در برابر سارکوم
از آنجا که MPNST تمایل زیادی به عود موضعی دارد، «پرتودرمانی» (Radiotherapy) تقریباً در تمامی موارد (بهویژه برای تومورهای بزرگتر از ۵ سانتیمتر) توصیه میشود. رادیوتراپی میتواند قبل از جراحی برای کوچک کردن تومور و تسهیل جراحی (Pre-operative) یا بعد از عمل برای پاکسازی سلولهای سرطانی احتمالی باقیمانده (Post-operative) انجام شود. استفاده از پروتکلهای نوین مانند «پرتودرمانی با شدت تعدیل شده» (IMRT) به پزشکان اجازه میدهد دوزهای بالایی از انرژی را مستقیماً به تومور بتابانند و آسیب به بافتهای سالم مجاور را به حداقل برسانند.
نقش «شیمیدرمانی» (Chemotherapy) در درمان MPNST کمی پیچیدهتر است. این تومورها به طور سنتی مقاومت نسبی به شیمیدرمانی نشان میدهند، اما در مواردی که سرطان به نقاط دوردست مثل ریهها گسترش یافته (متاستاز) یا زمانی که تومور اولیه بسیار بزرگ و غیرقابل جراحی است، از داروهای قدرتمندی مانند ایفوسفامید و دوکسوروبیسین استفاده میشود. تحقیقات فعلی بر روی «درمانهای هدفمند» (Targeted Therapy) متمرکز است که سعی دارند مسیرهای سیگنالدهی خاص در سلولهای NF1 را مسدود کنند تا رشد تومور را متوقف نمایند.
“
دانستنی نایاب:
برخلاف بسیاری از سارکومها، MPNST میتواند در آزمایشهای پاتولوژی نمایی شبیه به تومورهای مختلف داشته باشد؛ این پدیده را «تمایز هترولوگ» مینامند. در موارد نادری، بخشی از این تومور عصبی ممکن است شبیه به تومور عضلانی یا حتی غضروفی به نظر برسد که نشاندهنده ماهیت ناپایدار ژنتیکی آن است.
۷-توانبخشی عصبی؛ بازسازی مسیرهای حرکت
پس از اتمام درمانهای اصلی، سفر بیمار در مسیر «توانبخشی» (Rehabilitation) آغاز میشود. از آنجا که جراحی MPNST اغلب مستلزم آسیب به اعصاب محیطی است، بیمار ممکن است با چالشهایی نظیر افتادگی مچ پا (Foot drop)، ضعف در گرفتن اشیاء یا دردهای مزمن عصبی روبرو شود. «فیزیوتراپیستها» با طراحی برنامههای تمرینی اختصاصی، به تقویت عضلات باقیمانده و جلوگیری از تحلیل رفتن (Atrophy) آنها کمک میکنند. استفاده از بریسها و وسایل کمکی حرکتی میتواند استقلال بیمار را در فعالیتهای روزمره بازگرداند.
«کاردرمانی» (Occupational Therapy) نیز نقش کلیدی در بازیابی مهارتهای ظریف، بهویژه در تومورهای دست و بازو دارد. علاوه بر این، مدیریت دردهای پس از جراحی (Post-surgical pain) که گاهی به صورت دردهای فانتوم (حس درد در اندام قطع شده) یا دردهای نوروپاتیک شدید بروز میکند، نیازمند همکاری با متخصصان طب درد است. استفاده از تحریکات الکتریکی عصب (TENS) و داروهای تنظیمکننده عصبی میتواند کیفیت زندگی بیمار را در این دوران به شدت بهبود ببخشد.
۸-پایش بلندمدت و پیشآگهی؛ اهمیت مراقبتهای پس از درمان
به دلیل ماهیت تهاجمی MPNST، پایش (Follow-up) دقیق و مادامالعمر برای این بیماران الزامی است. در دو سال اول پس از درمان که بیشترین احتمال بازگشت وجود دارد، معاینات فیزیکی و تصویربرداریهای ریه و محل تومور هر ۳ تا ۶ ماه یکبار انجام میشود. پیشآگهی (Prognosis) بیماری به عوامل متعددی بستگی دارد که مهمترین آنها اندازه تومور در زمان تشخیص و امکان برداشتن کامل آن با حاشیه سالم است.
بیمارانی که تومور آنها کمتر از ۵ سانتیمتر بوده و به طور کامل جراحی شدهاند، شانس بقای بسیار بالاتری دارند. برای افراد مبتلا به NF1، این پایش شامل بررسی دقیق هرگونه تغییر در نوروفیبرومهای قدیمی است. ظهور دردی که شبها بیدار میکند، رشد سریع یک توده قدیمی یا تغییر در قوام توده، باید بلافاصله به عنوان یک هشدار جدی برای تبدیل شدن به بدخیمی تلقی شود. آگاهی عمومی و سواد سلامت در این زمینه، موثرترین ابزار برای تشخیص در مراحل قابل درمان است.
سوالات متداول (Smart FAQ)
نتیجهگیری
تومور بدخیم غلاف عصبی محیطی (MPNST)، سارکومی تهاجمی است که تشخیص و درمان آن نیازمند دقت وسواسی و همکاری تیمی متخصصان است. اگرچه پیوند این بیماری با ژنتیک و سوابق پرتودرمانی، مسیر مبارزه را دشوار میکند، اما پیشرفتهای شگرف در جراحیهای حفظ اندام، رادیوتراپی دقیق و توانبخشیهای نوین، چشمانداز بهبودی را برای بیماران روشنتر کرده است. کلید موفقیت در این نبرد، جدی گرفتن علائم عصبی، پایش مستمر در بیماران NF1 و مراجعه به مراکز تخصصی سارکوم است. با آگاهی، تشخیص زودهنگام و رویکردی جامع، میتوان با این بیماری مقابله کرد و کیفیت زندگی را به بازماندگان بازگرداند.
تجربه و نگاه شما ارزشمند است
مواجهه با بیماریهای نادری چون MPNST میتواند مسیری دشوار و گاهی تنهایی باشد. اگر شما یا عزیزانتان تجربهای در زمینه پایش ژنتیکی NF1 یا مسیر درمان سارکومهای عصبی دارید، دعوت میکنیم تجربیات خود را در بخش نظرات بنویسید؛ کلام شما میتواند برای دیگرانی که در ابتدای این راه هستند، مایه دلگرمی و آگاهی باشد.

بیش از دو دهه در زمینه سلامت، پزشکی، روانشناسی و جنبههای فرهنگی و اجتماعی آنها مینویسد و تلاش میکند دانش را ساده اما دقیق منتقل کند.
پزشکی دانشی پویا و همواره در حال تغییر است؛ بنابراین، محتوای این نوشته جایگزین ویزیت یا تشخیص پزشک نیست.
مطالب مرتبط