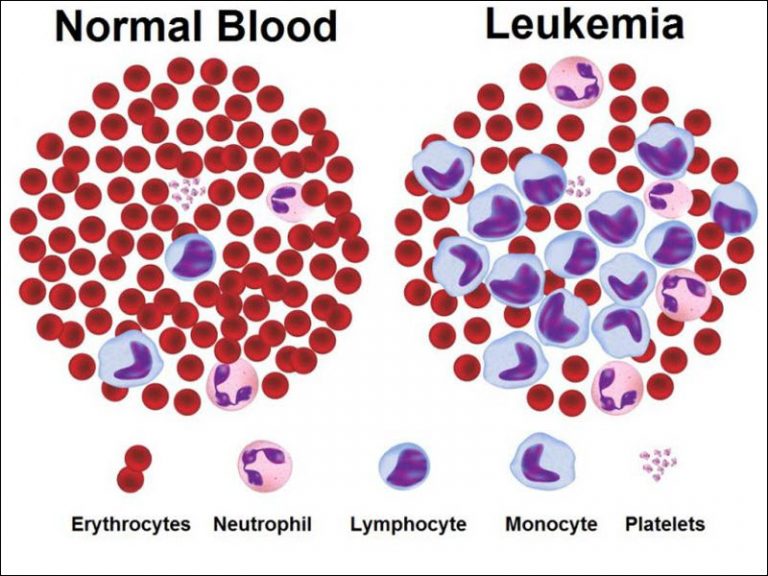
لوسمی میلوژن مزمن یا CML چیست؟ راههای تشخیص و درمان آن
لوسمی میلوژن مزمن (chronic myelogenous leukemia) یک بیماری پرولیفراتیو است که در آن عمدتاً ردهٔ سلولی گرانولوسیتی افزایش مییابد و همزمان با هیپرپلازی پلاکتها و ردهٔ اریتروئید همراه است. خصوصیت منحصر به فرد این بیماری در بین نئوپلاسمهای میلوپرولیفراتیو، سیر بالینی آن است که در تمامی موارد به لوسمی حاد تبدیل میشود. لوسمی میلوژن مزمن نخستین بیماری بدخیم خونی است که ارتباط آن با یک ناهنجاری کروموزومی اختصاصی به اثبات رسید. در بیش از 95% بیماران، گسترش کلونی نوعی سلول بنیادی دیده میشود که واجد کروموزوم فیلادلفیا شده است. در این حالت جابجایی متوازنی بین کروموزومهای 9 و 22 [11q: 34q][22؛9]t روی میدهد. این جابجایی، ژن abl (ویروس لوسمی آبلسون) بر روی کروموزوم 9 را به ژن bcr (ناحیهٔ خوشهٔ نقطهٔ شکست) (breakpoint cluster region) بر روی کروموزوم 22 متصل میکند و در نتیجه ژن سرطانزای جدیدی به نام bcr-abl ایجاد میشود. فراوردهٔ bcr-abl یک تیروزین کینازِ سیتوپلاسمی است که خود به خود فعال است و در سلولهای خونساز، لوسمی ایجاد میکند. پروتئین ادغامی bcr-abl گروهی از مکانیسمهای انتقال پیام را فعال میکند که به سلول اجازه میدهند مستقل از تنظیم سیتوکینها و تأثیر استرومای مغز استخوان، رشد کند. همچنین باعث میشود سلولهای CML نسبت به شیمیدرمانی مقاوم و در مقابل مرگ برنامهریزی شدهٔ سلولی (آپوپتوز) محافظت شوند. گروهی از بیمارانِ مبتلا به CML که فاقد کروموزوم فیلادلفیا هستند، دارای محصولات حاصل از اتصال bcr-abl بوده در واکنش زنجیره پلیمرازترانسکریپتاز (RT-PCR) میباشند که نشاندهنده ترانسلوکاسیون در سطح پایینتر از کروموزوم (ژنی) همراه با تولید همان فرآورده ژنی پاتولوژیک میباشد. تشخیص CML براساس شناسایی کروموزوم فیلادلفیا توسط روشهای کاریوتیپ، PCR و یا آنالیز هیبریدیزاسیون فلورسانت (FISH) صورت میگیرد. شناسایی کروموزوم فیلادلفیا امکان تشخیص راحتتر و پایش بیماری را فراهم کرده است. روشهای بسیار حساس و کمی RT-PCR امکان آن را فراهم ساخته است که نه تنها یک سلول مثبت از نظر bcr-abl در بین 105 تا 106 سلول خون محیطی شناسایی شود. بلکه وضعیت بیماری را هم از روی نمونههای خون محیطی و هم نمونه مغز استخوان میتوان تعیین کرد. امروزه پاسخ به رژیمهای درمانی در CML گزارش میشود و بین فروکشهای خونی (بازگشت تعداد گویچههای خون محیطی به حد طبیعی)، سیتوژنتیک (از بین رفتن کروموزوم فیلادلفیا) و مولکولی (کاهش لگاریتمی رونوشتهای bcr-abl قابل ردیابی تا کمتر از حد استاندارد توسط RT-PCR) افتراق قائل میشوند.
تظاهرات بالینی
لوسمی میلوژن مزمن، 20-15% تمامی موارد لوسمی را تشکیل میدهد و میزان بروز آن یک در هر صد هزار نفر جمعیت است. متوسط سن شروع 53 سالگی است اما بیماری میتواند هر ردهٔ سنی را مبتلا کند. حدود 40% بیماران در ابتدا بدون علامت هستند. سایر بیماران با خستگی، بیحالی، تنگی نفس، کاهش وزن، کبود شدن پوست و سیری زودرس مراجعه میکنند. معاینهٔ جسمانی معمولاً بزرگی طحال را نشان میدهد.
سیر طبیعی لوسمی میلوژن مزمن شامل مرحلهٔ مزمنی است که به بحران حاد بلاست تبدیل میشود. بیماران اصولاً در مرحلهٔ مزمن تشخیص داده میشوند که مرحلهٔ بی سروصدایی است و 3 تا 5 سال طول میکشد. در این مرحله، تعداد گویچههای سفید خون محیطی افزایش مییابد که همراه با ائوزینوفیلی و بازوفیلی میباشد (20%>)، اما تعداد بلاستهای خون ناچیز (5%>) است. با کنترل تعداد گویچههای خون محیطی این بیماران اساساً در طول این دوره بدون علامت هستند. نهایتاً بیماری وارد مرحلهٔ تسریع شدهای میشود که خصوصیات آن شامل تب، کاهش وزن، بزرگ شدن بیشتر طحال و درد استخوانی به دلیل سرعت بالای تولید و تخریب سلولهای مغز استخوان میباشند. کمکم علیرغم درمان، گویچههای سفید خون افزایش یافته و تعداد سلولهای بلاست در گردش خون (بین 19-10%) زیاد میشود. افزایش بازوفیلهای خون محیطی (20%<) منجر به تولید هیستامین همراه با علایم خارش، اسهال، و گرگرفتگی میباشد. در مرحلهٔ تسریع شده، بیمار ممکن است دچار اسپلنومگالی فزاینده، ترومبوسیتوپنی پایدار، یا ترومبوسیتوز و لکوسیتوز شود که با ناهنجاریهای سیتوژنتیک دودمانی جدید در مغز استخوان همراهاند.
مرحلهٔ آخر لوسمی میلوژن مزمن، بحران بلاست (blast crisis) نامیده میشود که نشان دهندهٔ تبدیل بیماری به لوسمی حاد است و طی آن بلاستها (20≤) مغز استخوان را اشغال میکنند و عناصر سلولی بالغ طبیعی در مغز استخوان و خون محیطی از بین میروند. مرگ ظرف چند هفته تا چند ماه روی میدهد. دو سوم بیماران دچار لوسمی میلوئید میشوند و در مابقی آنان لوسمی لنفوئید بروز میکند. یافتهٔ اخیر ثابت ثابت میکند که سلول نئوپلاستیک اولیه در این بیماری سلول بنیادی اولیهای است که قادر است به چندین رده تمایز یابد.
بررسیهای آزمایشگاهی
تغییرات آزمایشگاهی عبارتاند از: افزایش بارز شمار گویچههای سفید خون (به طور متوسط L. 109x 170) همراه با پایین بودن میزان فسفاتاز قلیایی، بالا بودن میزان اسید اوریک و لاکتات دهیدروژناز و ترومبوسیتوز، بررسی گسترهٔ خون محیطی در CML فاز مزمن، مجموعهٔ کاملی از سلولهای میلوئید را در تمام مراحل رشد گرانولوسیتی نشان میدهد که شامل میلوبلاستهای نابالغ (معمولاً کمتر از 5%)، میلوسیتها متامیلوسیتها، بازوفیلها، ائوزینوفیلها، سلولهای باند، و نوتروفیلها میباشند. در مقابل، گسترهٔ خون محیطی در هیپرپلازیهای گرانولوسیتی واکنشی (واکنش لوکموئید) که ناشی از عفونت یا سپسیس حاد هستند، عمدتاً شامل نوتروفیلهای بالغ و سلولهای باند همراه با تعداد اندکی میلوسیت، بازوفیل، یا ائوزینوفیل میباشد. مغز استخوان در CML، پرسلول و متراکم است و تعداد سلولهای میلوئید در تمام مراحل به طرز چشمگیری افزایش یافته و فیبروز رتیکولین دیده میشود. شناسایی کرموزوم فیلادلفیا و یا رونوشتهای غیرطبیعی bcr-abl با آزمونهای مرسوم یا ملکولی، تشخیص CML؛ را تأیید میکند.
درمان
در گذشته، داروهای شیمیدرمانی خوراکی نظیر هیدروکسی اوره و بوسولفان در کاهش تعداد سلولهای میلوئید در مرحلهٔ مزمن CML مورد استفاده بودند. هر چند این اقدام عوارض حاد بیماری را کاهش میداد اما تأثیری بر روی پیشآگهی طولانی مدت بیماری یا پیشگیری از ورود به بحران بلاست نداشت. استفاده از اینترفرون آلفا سبب همان میزان فروکشهای خونی (80-60%) موارد میشود و اولین دارویی بوده است که منجر به پاسخهای سیتوژنتیک در 30-20% از این بیماران میشود. نشان داده شده است که ترکیب شیمیدرمانی با اینترفرون سبب افزایش بیشتر میزان پاسخهای سیتوژنتیک میگردد. پاسخهای سیتوژنتیک ناشی از رژیمهای حاوی اینترفرون با افزایش طول عمر بیماران همراه است. بیماران تحت درمان با اینترفرون آلفا همچنان دارای سلولهایی با جابجایی bcr-abl هستند که با واکنش زنجیرهٔ پلیمراز قابل شناسایی است و به همین دلیل خطر عود بیماری در افراد همچنان وجود دارد. با این حال، بسیاری از بیماران علیرغم بیماری مولکولی قابل شناسایی، سالها در حالت فروکش خون و سیتوژنتیک باقی میمانند. مکانیسم کنترل بیماری با این رژیمهای دارویی علیرغم سلولهای مثبت bcr-abl معلوم نیست. بیمارانی که وارد مرحلهٔ تسریع شده و یا بحران بلاست میشوند، پاسخ ضعیفی به اینترفرون و مقادیر بالای شیمیدرمانی میدهند و پاسخها فقط موقتی و به مدت کمتر از 6 ماه میباشند.
معرفی داروی ایماتینیب مسیلات (Gleevec که سابقاً STI-571 خوانده میشد) برای درمان CML، نویدبخش نخستین درمان موفق هدفمند برای سرطان بود. گلیوک دارویی است که براساس منطق علمی طراحی شده و مهارکنندهٔ رقابتی عامل رشد مشتق از پلاکت، bcr-abl و کینازهای گیرندهٔ تیروزینی c-kit است. مهار فسفریلاسیون bar-abl منجر به توقف پیامدهی پایین دست و مسیرهای رشد شده و آپوپتوز سلولهای مثبت از نظر bcr-abl را القا میکند. مطالعات آزمایشگاهی نشان داده گلیوک میتواند با قدرت، رشد ردههای سلولی CML و سلولهای اجدادی که واجد bcr-abl هستند را مهار کند و میزان بقا را در مدلهای حیوانی تومور افزایش دهد. در سال 1998، نخستین آزمایههای بالینی با این داروی خوراکی مهارکنندهٔ تیروزین کیناز، روی بیمارانی که به اینترفرون آلفا پاسخ نداده بودند، آغاز گردید. نه تنها دارو به خوبی تحمل شد و عوارض جانبی قابل کنترل بودند، بلکه 96% بیمارانی که دوز بیش از mg 300 برای 4 هفته دریافت کردند وارد فروکشی (remission) هماتولوژیک شدند و 33% پس از 8 هفته وارد فروکشی سیتوژنتیک شدند. این نتایج تکاندهنده، در چندین کارآزمایی تأیید شد، به این ترتیب نشان داده شد گلیوک بهتر از اینترفرون آلفا و سیتارابین میتواند در بیماران درمان نشدهٔ جدیدی که در مرحلهٔ مزمن CML هستند موجب القای فروکشی شود. در یک پیگیری با میانگین 5 سال، درمان اولیه با گلیوک منجر به پاسخهای سیتوژنتیک کامل در 87% بیماران CML فاز مزمن با یک بقای کلی 89% تخمین زده شد. تنها 7% بیماران وارد CML فاز سریع شده و یا بلاست شدند و هم میزان پاسخ و نتایج معکوس درمان گلیوک با گذشت زمان درمان کاهش پیدا کرد. هیچ بیماری که پس از درمان گلیوک به مدت 12 تا 18 ماه به یک پاسخ کامل سیتوژنتیک و فروکشی مولکولی دست پیدا کرده بود (که به صورت یک کاهش لگاریتمی سه در رونوشتهای bcr-abl تعریف میشود)، پس از پنج سال دچار پیشرفت بیماری نشد. براساس این مطالعه، درمان گلیوک اکنون رمان استاندارد برای CML در فاز مزمن میباشد. با وجود این نتایج، این عامل بیماری را درمان قطعی نمیکند.
همانند مشکلی که در درمان با اینترفرون وجود داشت، اکثر بیمارانی که با مصرف گلیوک به CCR میرسند، باز هم در تستهای حساس ملکولی، وجود سلولهای بنیادی CML را که bcr-abl مثبت هستند، نشان میدهند. بنابراین برای کنترل بیماری، باید گلیوک تا پایان عمر مصرف شود، و حتی بیماران با کنترل کامل CML فاز کرونیک روی درمان با گلیوک همچنان در ریسک پیشرفت نهایی بیماری و شکست درمان هستند. با وجود آنکه دوزهای بالای گلیوک میتواند پاسخهای هماتولوژیک و سیتوژنتیک گذرا در بیمارانی با CML فاز تسریع شده و بلاستی را القا کند، این بیماران در بهترین حالت کوتاه مدت هستند، و مقاومت به گلیوک در مراحل پیشرفتهٔ بیماری ثابت شده است. در نیمی از بیماران با مقاومت بالینی به گلیوک جهشهای تک نوکلئویتدی در ژن bcr-abl منجر به تغییرات ساختاری در کیناز bcr-abl میشود که اتصال دارد و بنابراین اثرات مهاری آن را تغییر میدهد. در نهایت، درصد کمی از بیماران CML قادر به تحمل اثرات جانبی گلیوک، که عمدتاً در دستگاه گوارش هستند، نمیباشند. برای حل این موضوع، مهار کنندههای جدیدتر کیناز bcr-abl ساخته شدهاند، به طور ویژه dastainib (sprycel) و nilotinib (Tasigna) برای درمان بیماران CML مقارم یا حساس به گلیوک.
هر دو اینها در مقایسه با گلیوک قدرت بیشتری را در مقابل کیناز bcr-abl در محیط آزمایشگاه نشان دادهاند و در سلولهای CML بیاان کنندهٔ اکثر جهشهای bcr-abl شناخته شده از خود فعالیت نشان دادهاند. تجربیات بالینی در فاز یک با تک – مادهٔ داستینیب یا نیلوتینیب در بیماران که مبتلا از درمان با ایماتینیت شکست خوردهاند، به طرز ثابتی سبب پاسخهای سیتوژنتیک در نزدیک به 40% از بیماران شده است. با این وجود، فقدان تجربیات بالینی که این دو ماده را مستقیماً مقایسه کنند، و نیاز به منتظر بودن برای دادههای نتایج بالینی دراز مدت برای هر یک از دو دارو، امکان انتخاب یک مهار کنندهٔ bcr-abl خط دوم را برای بیماران منفرد و یا پزشکان از بین میبرد. شرح حال مسمومیت هر دو دارو به فراوانی عدم همپوشانی با گلیوک را نشان میدهد.
در حال حاضر، بیش از شروع استفاده گسترده از درمان هدفدار مهار کنندهٔ کیناز bcr-abl ریشه کنی تمامی سلولهایی که مقادیر قابل شناسایی از جابجایی bcr-abl داشته باشند، فقط پس از پیوند آلوژنیک سلول بنیادی امکانپذیر است. پیش از شروع استفاده گسترده از درمان هدفدار مهار کنندهٔ کیناز bcr-abl به بیماران جوانی که در مرحلهٔ مزمن CML بودند، در بدو تشخیص پیوند آلوژنیک مغز استخوان از یک دهندهٔ سازگار (از نظر HLA) به عنوان درمان قطعی پیشنهاد میشد. در دراز مدت، میانگین بقای این دسته از بیماران پس از پیوند، معادل 75-50% بود. به دلایل ناشناخته، در بیمارانی که ظرف یک سال پیش از تشخیص تحت این درمان قرار میگیرند نتیجهٔ نهایی پیوند بهتر از سایر موارد است. شواهد فزایندهای حاکی از آن است که پاسخ عالی بیماران مبتلا به CML به پیوندِ سلول بنیادی، تا حدودی مربوط به سرکوب فعال بیماری توسط سلولهای پیوندی جدید است که این حالت، اثر پیوند علیه لوسمی (GVL) graft-versus-leukemia effect نامیده میشود. با این وجود، کنترل عالی و مسمومیت کلی پایین مهار کنندههای bcr-abl در دراز مدت برای CML فاز مزمن در مقایسه با عوارض و مرگ و میر 20% تا 30% پس از SCT، به طرز مؤثر SCT را به صورت یک گزینهٔ درمانی تنها برای آن گروه از بیماران CML فاز مزمن که مستقیماً در مقابل گلیوک شکست میخورند، تبدیل کرده است (مثلاً بحران بلاست، عدم تحمل). تنها استثنا در این مورد، افرادی با بیماری CML بیان کنندهٔ جهشهای کیناز bcr-abl (همانند T3.51) هستند که به تمامی درمانهای مهار کنندهٔ bcr-abl مقاوم شناخته شدهاند. این بیماران باید در مراحل اولیهٔ روند بیماری شناخته شوند تا از نظر درمان قطعی SCT در مقابل درمان با گزینههای تجربی مورد توجه قرار گیرند. پیوند همچنین به صورت تنها گزینهٔ درمانی قطعی در CML با سن بالا باقی مانده است. بیماران با CML فاز سریع شده ممکن است در حین انتظار برای پیوند آلوژنیک (در صورت امکان) تحت درمان با گلیوک با دوز بالا و یا درمانهای تجربی قرار گیرند، در حالیکه آنهایی که دو فاز بلاست قرار دارند باید تحت شیمیدرمانی القایی براساس رژیمهای لوسمی حاد قرار گیرند که با پیوند و یا آزمایش بالینی پیگیری خواهد شد.
به طور کلی دگرگون شدن CML، از یک سرطان پیشروندهٔ کشنده به سرطانی که در آن تقریباً 90% بیماران با یک بیماری پایدار و درمان خوراکی کیناز که پس از 5 سال زنده هستند، یکی از دستآوردهای افتخارآمیز در درمان سرطان در دههٔ گذشته میباشد. میانگین بقای کلی بیماران CML به طرز چشمگیری از چند ماه به چند سال (در بیماری اول قرن 20 افزایش پیدا کرده است) و در بیماران تحت درمان با اینترفرون تا 6 سال و هنوز دو مورد درمان مهار کنندهٔ bcr-abl تعیین نشده است. با این وجود، جست و جو برای یک درمان قطعی بیماری cml و نیز برای درمان بهتر آن گروه از بیماران CML که بیماری مقاوم به گلیوک دارند، یک چالش درمانی برای سالهای پیشرو محسوب میشود.
مطالب مرتبط